
Abstract
Secretory and transmembrane proteins undergo post-translational modifications and folding in the subcellular organelle, that is, endoplasmic reticulum (ER) to become functionally active. Various factors such as high oxidative stress, low glucose, calcium imbalance, and viral infections interfere with the ER protein folding functions, leading to accumulation of unfolded and misfolded proteins that activate downstream signal transduction pathways, termed as unfolded protein response (UPR). This UPR signaling is adaptive and restored the normal function of cells by decreasing protein synthesis, increasing the folding capacity of ER and degradation of misfolded proteins. If the stress condition is overwhelmed, then UPR signaling shifts to apoptotic pathways. However, cancer cells utilized these UPR signaling for their survival and progression as an adaptive mechanism. In this review, the authors discuss about the overview of ER stress and subsequent UPR signaling and various aspects of cancer as survival, proliferation, and angiogenesis in relation to UPR. Understanding the UPR signaling in relation to cancer will be further helpful in designing therapeutics against cancer.
Introduction
Endoplasmic reticulum (ER) is the subcellular organelle that involved in the synthesis and folding of secreted and transmembrane proteins, calcium regulation, and lipid biosynthesis. Properly folded proteins are targeted to their destined site whereas the terminally misfolded proteins are subjected to degradation by the ER-associated degradation pathways. The oxidative environment of the ER is essential for disulfide bond formation and proper folding of the secretory proteins. As the ER is the site of intracellular calcium storage, many calcium-dependent molecular chaperones as glucose-regulated proteins (GRP78, GRP94), calreticulin, etc., are associated with the protein folding, stabilization of the intermediates, and the post-translational modifications. Under normal conditions, there is balance between the level of folded and misfolded proteins but under stress conditions the functioning of ER is disrupted that leads to the accumulation of misfolded and unfolded proteins in the ER lumen. The disturbance in the folding machinery leads to the increased accumulation of misfolded and unfolded proteins beyond the handling capacity of ER, resulting in ER stress. Subsequently, this stress triggers the activation of downstream signal transduction response that is collectively termed as unfolded protein response (UPR). These signaling cascades, in turn, activate the expression of the genes that maintain or bring down the homeostasis in the ER functioning or if necessary kill the cell. 1 –4 Many physiological conditions such as glucose deprivation, calcium imbalance, hypoxia, increased reactive oxygen species level, and free fatty acids, and pathological conditions such as viral infection are responsible for triggering ER stress. 5 Glucose deprivation induces ER stress by interfering with N-linked glycosylation. 6 Various chaperones activity is dependent on calcium, thus disturbance in calcium regulation and concentration also hinders the protein folding process and leads to the accumulation of unfolded proteins. 7 Viral infection is also responsible for ER stress generation and destined the cells to apoptosis. 8,9 Some of the basal misfolded proteins are degraded by the proteasome, so the impairment of proteosomal functions also results in misfolded protein aggregates and pathogenesis of various neurodegenerative diseases and cancer. 10,11
The role of ER stress and UPR pathways is known in many types of cancer. The redox and nutrient status, increased mutational load, and high protein need induce ER stress in the cancer cells. 12 –14 The ER stress subsequently activates UPR pathways, which confer survival, growth, and chemoresistance. 15 –17 Activation of UPR under stressed environment of tumor is essential for oncogenic growth and survival. The UPR components also contribute to tumor progression by interacting with many oncogenes. The adaptive benefits of UPRs in the cancer cells further complicate the use of chemotherapeutics against cancer.
In this review, the authors discuss about the ER stress and UPR pathways as a whole in normal cells and also their role in tumor progression and survival, which can be beneficial to understand the UPR functioning under tumor microenvironment and designing therapeutics for cancer treatment.
Unfolded Protein Response
Accumulated unfolded and misfolded proteins in the ER lumen activate the transmembrane ER stress sensors, which initiate signal transduction pathways termed as UPR. The three arms of the UPRs are inositol-requiring kinase/endoribonuclease 1 (IRE-1), activating transcription factor 6 (ATF6), and protein kinase activated by double-stranded RNA-like ER kinase (PERK) (Fig. 1). 18,19 These ER transmembrane proteins are generally in inactive form by binding with the ER chaperones such as GRP78. GRP78 holds the N-terminal of these transmembrane proteins, preventing their activation. Increased burden of unfolded and misfolded proteins activates them and mediates the downstream signaling.
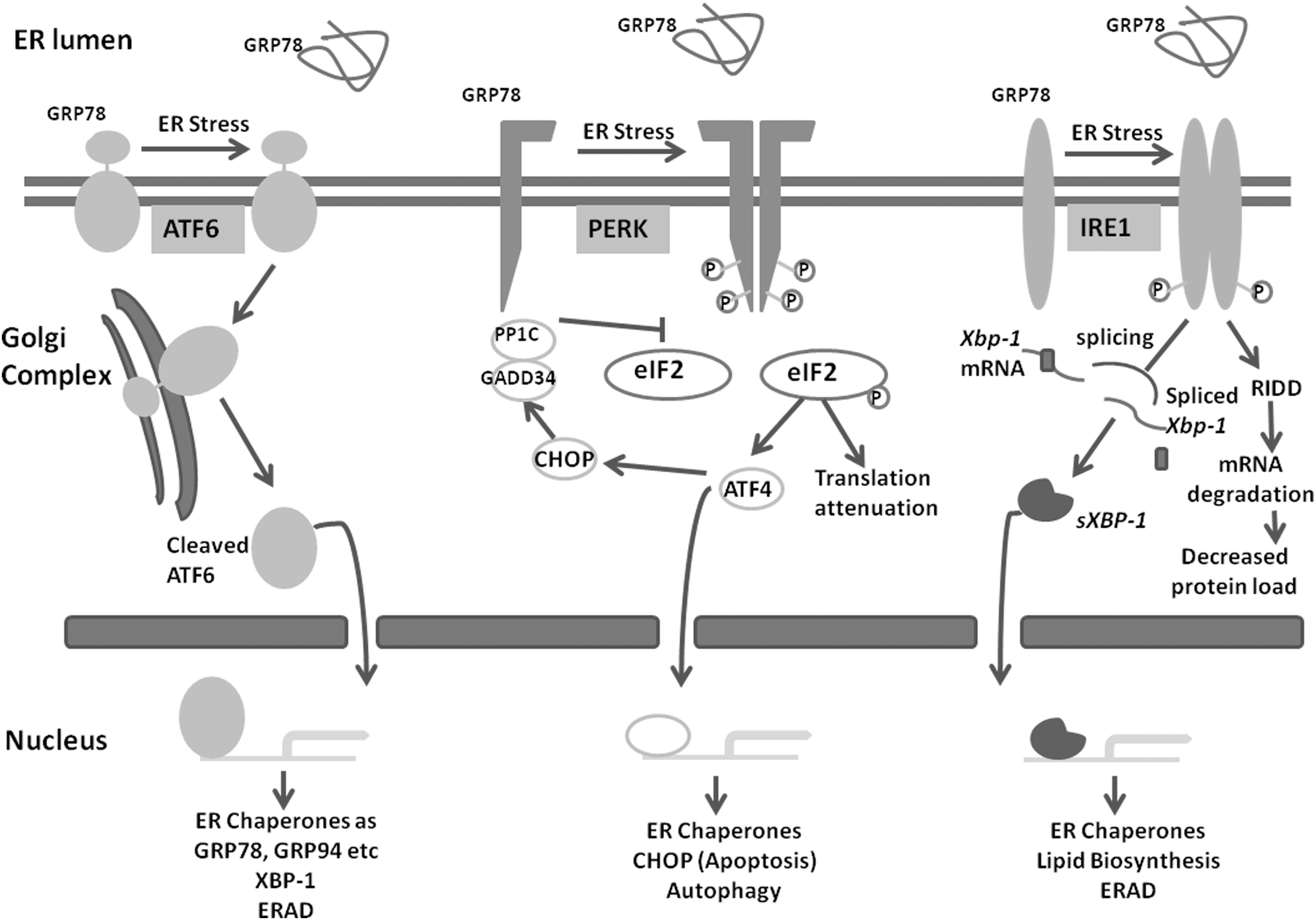
The three arms of unfolded protein response, namely, IRE-1, ATF6, and PERK become activated under accumulation of misfolded and unfolded proteins in the ER lumen. These downstream signaling molecules restore the ER homeostasis by increasing the expression of chaperones, which assist in protein folding, by attenuating translation, and by degrading mRNA to decrease protein load in the ER. Under high level and persistent stress, these signaling destined the cells to apoptosis (CHOP, JNK pathway). ATF4, activating transcription factor 4; ATF6, activating transcription factor 6; CHOP, CCAAT-enhancer-binding protein homologous protein; eIF2, eukaryotic translational initiation factor-2; ER, endoplasmic reticulum; ERAD, endoplasmic reticulum-associated degradation; GADD34, growth arrest and DNA-damage-inducible 34; GRP78, glucose-regulated protein 78; GRP94, glucose-regulated protein 94; IRE-1, inositol-requiring kinase/endoribonuclease 1; PERK, protein kinase activated by double-stranded RNA-like ER kinase; RIDD, regulated IRE-1-dependent decay.
IRE-1
IRE-1 is the most conserved UPR signaling pathway also present in lower eukaryotes. 20 PERK and ATF6 pathways were later evolved in metazoans. IRE-1 is an ER transmembrane protein with a luminal domain for sensing the ER stress environment and a cytoplasmic domain with protein kinase activity. In response to direct binding of the unfolded proteins, the IRE-1 becomes oligomerized and transautophosphorylates the cytoplasmic domain. In this pathway, there is no sequential activation of protein kinases, rather the activated phosphorylated cytoplasmic domain causes site-specific endoribonuclic cleavage of xbp-1 mRNA. Thus, the cytoplasmic domain of IRE-1 has dual role with kinase and endoribonuclease activity. The splicing thus occurs by the nonconventional mode. Spliced XBP-1 (sXBP-1) translocates to the nucleus where it acts as transcriptional activator of UPR target genes. Spliced and unspliced products have different roles in the activation of UPR genes. 19,21 Spliced XBP-1 is more stable than the unspliced form. Spliced XBP-1 mainly regulates the lipid biosynthesis, expression of chaperones, and endoplasmic reticulum-associated degradation (ERAD) components. 2,22,23 The IRE-1 pathway increases the protein folding environment in the ER lumen. The IRE-1 branch also reduces the protein load on the ER by degrading some of the mRNA by its endoribonuclease activity, which is termed as IRE-1-mediated mRNA decay (regulated IRE-1-dependent decay [RIDD]). 19,24
ATF6
ATF6 is a metazoan-specific ER stress sensor, present as inactive precursors. ATF6 is an ER transmembrane protein that senses the unfolded protein status in the ER lumen by the luminal domain, becoming activated and transported to the Golgi body by packaging into the transport vesicles. In the Golgi body, it is cleaved sequentially by the two proteins site1 protease and site 2 protease (S1P and S2P). 25,26 The cleaved N-terminal cytoplasmic domain then translocates to the nucleus and regulates the transcription of UPR target genes. 27 This pathway increases the protein folding capacity of ER by activating the expression of the chaperones. The primary targets of ATF6 are BiP (GRP78, a chaperone involved in protein folding), GRP94, protein disulphide isomerise, etc. It is a long-term adaption to restore ER homeostasis. 1,2
PERK
PERK, an ER-resident transmembrane kinase, is phylogenetically related to IRE-1. It is also a type 1 transmembrane protein like IRE-1. Its cytoplasmic domain with protein kinase activity is activated by oligomerization and then trans-autophosphorylated. Unlike IRE-1 wherein there is no downstream substrate for phosphorylation, in the PERK pathway, sequential downstream phosphorylation cascade is activated. Eukaryotic translational initiation factor-2 (eIF2) is phosphorylated at ser51 by activated PERK cytoplasmic domain, which inhibits its further activation by replacing active GTP with guanine nucleotide exchange factor eIF2B. Inactivation of eIF2 results in lower translational initiation, ultimately reducing total protein synthesis and decreasing the protein folding burden on the already stressed ER. PERK pathway not only reduces ER load but also has a role in transcriptional activation of UPR genes. However, in PERK pathway activation, when eIF2 is limiting, some mRNAs with short open reading frame (ORF) in their 5′UTR-like ATF4 are preferentially translated. Increased ATF4 then transcriptionally increases the expression of CHOP (transcription factor CCAAT-enhancer-binding protein homologous protein) and growth arrest and DNA-damage-inducible 34 (GADD34), which, in turn, involves in the apoptosis. 19,28 –30 GADD34, whose expression is increased under high stress, in negative feedback program dephosphorylates eIF2. Thus at the moderate level of stress, PERK pathway is protective but can drive the cells toward apoptosis if stress is prolonged.
The initial signaling of UPR pathways adopts to restore the normal function of ER by altering the expression of genes to increase protein folding capacity, translational attenuation to decrease ER protein folding load, and removal of misfolded proteins by ERAD pathway. If the ER stress is persistent and cannot be mitigated, then prolonged activation of UPRs leads to cell death pathways. 19,31
ER Stress: Cell Survival or Death Decision
The three branches of the UPRs are activated independently by different transmembrane receptors in the ER membrane but they are interconnected and mediate the cellular response to align the cellular physiology and restore the ER functioning. Activation of the UPRs also affects cell metabolism. These signaling events selectively increase the expression of ER chaperones involved in protein folding. 2 In the case of prolonged and unmitigated ER stress, the cells are not able to restore homeostasis which results in the cell death. The decision between cell survival and death depends mainly on the duration and intensity of the stress exerted or exposed. The mechanism is unclear when cell switches to apoptosis under stress. All the three branches of UPR initially oppose the death signals but sustained stress shifts the cytoprotection toward apoptosis or cell death. 1,19 Under the persistent ER stress, the IRE-1 signaling becomes attenuated. 32 PERK pathway is also deactivated as eIF2-P is dephosphorylated by GADD34, which is transcriptionally activated by CHOP. These two pathways as an intrinsic timer decide between life and death. Chronic activation of UPR pathways not only shifts from survival to death but also alters cell function as seen in chondrocytes. 33 In the case when cells cannot overcome the stress, altered calcium concentration leads to the activation of death effectors Bak and Bax by translocation from ER to mitochondria, 34 and also activates caspase 12. 35,36 Survival pathways under low level of stress are attributed to the low stability of CHOP and GADD34. Under chronic and severe ER stress, the expression of these proteins exceeds, leading to death. 37 eIF2 phosphorylation by PERK pathway plays the major role in deciding cell survival or death under stress. PERK phosphorylates and inactivates eIF2, globally decreasing the protein synthesis under ER stress. Owing to the presence of short ORF in 5′UTR of ATF4, phosphorylation of eIF2 favors its expression, which is a transcription factor for CHOP. CHOP then activates GADD34, which along with protein phosphatase 1 dephosphorylates eIF2. Thus, eIF2 phosphorylation contributes to cell death by increasing expression of cell death mediators as CHOP, 38 which further represses BCL-2 (an antiapoptotic) expression. 39 This apoptosis is necessary to protect surrounding cells from the rogue cells with defective signaling component. In C. elegans, mutation in either IRE-1 or ATF6 branch is tolerated but compromising both the IRE-1 and ATF6 branch hinders the worm development. 40 The three branches of UPR are activated independently; however, they cross-talk with each other and the outcome of the these pathways overlaps significantly. 2
UPR Signaling in Cancer
ER stress is linked with many pathophysiological conditions. The high demand of proteins and mutations in the highly proliferative tumor cells affects the protein folding process and thus induces the ER stress. 41,42 The tumor microenvironment is challenged with hypoxia, glucose deprivation, and high oxidative stress environment. Cancer cells have the capabilities to withstand the elevated oxidative stress, increased protein demand, and nutritional stress. Survival of tumor cells under this stressed environment depends on the downstream UPR signaling, which maintains proper ER functioning. 43 –45 Under ER stress, the UPR signaling is generally an adaptive strategy but can also lead to cell death if cells are not able to overcome the stress conditions. Some of the cancer types can bypass the UPR-induced death response and use the UPR signaling as a selective adaptive advantage only. 46 Tumor cells used these UPR pathways for their survival, angiogenesis, and cell proliferation, and avoid apoptosis and chemoresistance. For maintaining malignancy, tumor cells transmit the UPR signaling in surrounding endothelial cells and dendritic cells to induce angiogenesis, which is termed as transmissible ER stress. 47 There are many reports on UPR contribution in tumor survival and protection of cells from death. 16,17 The mechanism by which UPR signaling promotes cancer progression is by increasing the protein folding capacity to meet the high demands of proteins for proliferation. In normal cells under persistent stress, the IRE-1 and ATF6 pathways attenuate and PERK pathways induce apoptosis through CHOP. In some of the cancers, the ER stress-induced death can be overwhelmed by constitutive activation of the IRE-1–XBP-1 pathway. 48,49 The increased level of GRP78 also contributes for antiapoptotic phenotypes in many cancer types. In some of the tumors, high level of ER stress induces death of cells. 46,50 Hypoxia in solid tumor activates ATF4 and its downstream targets. 51,52 There are some reports wherein inhibition of ER stress induction is beneficial for the proliferation and progression of cancers such as multiple myeloma, breast cancer, and pancreatic cancer. 53 –55 Thus, the understanding of which type of tumors depends on the UPR for survival and which type of tumors is sensitive to chronic stress will be a future area of research in the development of cancer therapeutics.
Role of IRE-1 branch in cancer cell survival
IRE-1 branch activation after ER stress has a major role in cancer cell survival and progression. Various cancer types (ovary, lung, kidney, and brain) are associated with the mutation in the IRE-1 and its downstream targets. 56,57 Under low level of stress, IRE-1 branch confers the survival over death, but under prolonged stress condition, it can also draw the cells toward the apoptotic fate by recruiting TRAF2 and activating proapoptotic JNK2. To overcome these death signals, the cancer cells alter this arm of signaling by mutation in TRAF2. 19 The IRE-1 downstream targets, that is, XBP-1, also contributes to cancer cell survival and progression as its expression was found to be upregulated in many cancer tissues (breast, lung, and pancreas) 58,59 and its downregulation negatively affects the tumor growth and survival. 60 Role of IRE-1 branch of UPR is well known in multiple myeloma as it is required in mature B cell differentiation. 21,61,62 IRE-1 and XBP-1 mutations are well reported in multiple myeloma 63,64 and other types of cancers. 56,65
Role of PERK branch in cancer cell survival
PERK pathway in tumor cells either facilitates their survival or suppresses their progression. PERK branch confers oncogenic transformation by promoting myc-induced autophagic pathways. 66,67 CHOP (an apoptosis mediator) suppresses the tumor progression thus chop deletion has tumor progression phenotype in lung cancer. 13
Role of ATF6 branch in cancer cell survival
ATF6 branch has tumor progression role in hepatic cancers. 68,69 GRP78, which is downstream target of ATF6, showed increased expression in many malignant cells. In the tumor cells the increased level of GRP78 localized at the cell surface along with the ER lumen. 70 Cells with partial knockdown of GRP78 are unable to grow like a tumor in nude mice. 71
UPR and Angiogenesis
The other mean to support cancer cell survival and progression is through increasing the vascularization and blood supply to the proliferating cells known as angiogenesis, which, in turn, facilitates the supply of more nutrients and oxygen to the glucose-deprived and hypoxic cancer cells. UPR target genes play a significant role in the induction of the angiogenesis process in the tumor microenvironment. The main proangiogenic factor, that is, vascular endothelial growth factor (VEGF), is positively regulated by the UPR signaling for promoting angiogenesis in the cancer cells. 72,73 IRE-1 mutation inhibits angiogenesis that can be restored by expressing spliced XBP-1. 74,75 IRE-1–XBP-1 branch has the role in endothelial cell proliferation through Akt and GSkβ pathways. 76 UPR through its IRE-1 branch also has a role in embryogenesis and development by regulating the placental angiogenesis process. 62,77 GRP78 expression increased during early proliferative and the secretory phase of the menstrual cycle, which suggests that UPRs also have a role in the process of angiogenesis of endometrium tissues. 78 Induction of UPR due to increased metabolic rate in the cancer enhances the level of ATF4 and XBP-1 (downstream targets of IRE-1), which transcriptionally upregulates the expression of proangiogenic factors such as VEGF. 79,80 VEGF that is a major regulator of angiogenesis gets upregulated under hypoxic condition and also by the two transcription factors, that is, HIF-1α and HIF-1-β. 81,82 Besides this hypoxia, induction of UPR in cancer tissues also enhances the expression of VEGF by the downstream effectors of the IRE-1 branch, ATF4 and XBP-1, which directly bind to the VEGF promoter. 73,83 VEGF expression level is regulated by UPR at various stages that are independent of hypoxia-induced signaling in the tumor. Although UPR in the tumor cells is also activated under hypoxic condition, its role in angiogenesis and VEGF upregulation is mediated through the IRE-1 branch. Thus, upon induction of ER stress, various proangiogenic factors get upregulated and antiangiogenic factors downregulated in the tumor cells, which increase blood supply for the high demand of O2 and nutrients.
Targeting ER Stress as Therapeutic Implication in Cancer
Many pathological complications are associated with the disturbance in the protein folding machinery and concurrent ER stress. Various therapeutic drugs are known that can induce ER stress and destined the cells to apoptosis. 84 It is possible that an optimum level of ER stress and UPR signaling that is induced under tumor microenvironment is necessary for cancer survival and progression, but exacerbating the stress level beyond the handling capacity can drive the cancer cells toward death. UPR signaling can be targeted in cancer treatment in both ways, that is, either by inducing a high level of stress in the pre-existing stress microenvironment of tumor or by preventing activation of UPR that is beneficial for cancer progression and drug resistance, making them sensitive to other chemotherapeutic drugs. Many naturally occurring compounds including alkaloids, polyphenol, and saponins can be used to target ER stress for the medical benefits. 85 Many drugs or inhibitors of various steps of UPRs can be used in therapeutics in combination to existing cancer chemotherapy. The effect of various ER stress inducers in cancer therapy depends upon the type of cancer, thus further understanding the different types of tumor metabolism and its correlation with UPRs will be helpful for designing drugs against cancers.
Conclusions
In the secretory cells, the correctly folded proteins are translocated from the ER to the cell surface or outside the cells. Various physiological and pathological conditions disturb the ER functioning and the unfolded and misfolded proteins get accumulated in its lumen. To reduce the burden of these unfolded proteins, cells activate downstream UPR pathways to overcome ER stress and cell survival. There are different adaptive mechanisms such as repression of the translational machinery, transcriptional upregulation of ER chaperones, and ERAD. IRE-1 (through RIDD) and PERK (through eIF2-P) decrease the ER stress burden by reducing the amount of proteins that is synthesized and entered in the ER. 1,19,86 In case of prolonged and chronic ER stress, the UPR pathways lead to death of cells by other signaling cascades.
Various physiological challenges in the cancer cells such as hypoxia, low glucose, and nutritional stress directly interfere with the function of ER and concurrently lead to unfolded protein accumulation. Hypoxic conditions affect the disulphide bond formation thus affect the protein folding process and contribute to ER stress induction. 87 Various stress conditions in the microenvironment of tumors increase the protein folding load in the ER, which subsequently generates the ER stress. Cancer cells activate UPR by handling stress and conferring survival over death. Modulation or interfering with the UPR genes can be looked as potential targets to treat cancer or to make cancer chemotherapy more effective. 88 –90 There is a need to look forward for combinational therapy of existing chemotherapeutics along with UPR signaling targeting drugs as a promising approach for cancer treatment.
Footnotes
Disclosure Statement
No competing financial interests exist.