Synthesis and Evaluation of 2-[ 18 F]Fluoroethyltriazolesuberohydroxamine Acid for Histone Deacetylase in a Tumor Model as a Positron Emission Tomography Radiotracer
Available accessResearch articleFirst published online March, 2018
Synthesis and Evaluation of 2-[ 18 F]Fluoroethyltriazolesuberohydroxamine Acid for Histone Deacetylase in a Tumor Model as a Positron Emission Tomography Radiotracer
Histone deacetylases (HDACs) are an important regulator of expression and activity of numerous proteins in terms of epigenetic aberrations. This makes HDACs attractive for antitumor therapy and imaging in certain cancers. The authors report the radiochemical synthesis of 2-[18F]fluoroethyltriazolesuberohydroxamine acid ([18F]FETSAHA) as a HDAC-targeted radiolabel probe for positron imaging tomography/computed tomography. The authors also evaluated the in vivo tumor targeting in subcutaneously implanted RR1022 rats. [18F]FETSAHA was produced in less than 2 h with 31.2% ± 4.6% (n = 6) decay-corrected yields and specific activity of 21.4 ± 9.1 GBq/μmol (n = 6) at end of synthesis. [18F]FETSAHA showed significant radioactivity accumulation in tumors with rapid blood clearance and both gastrointestinal track and renal excretion. Tumor-to-blood and tumor-to-muscle uptake ratios in the RR1022 tumor bearing rat model were 1.21 and 1.83 and 2.75 and 2.76 at 30 and 60 min, respectively. An inhibition study of [18F]FETSAHA in the presence of excess amount of suberanilohydroximic acid (SAHA) revealed receptor specific activity accumulation. [18F]FETSAHA has favorable in vivo tumor imaging properties and may be useful for noninvasive evaluation of the correlation between cancer and HDACs.
Introduction
Human diseases, including cancer, have been regarded primarily as genetic disorders. However, epigenetic aberrations and genetic alterations are recognized as main factors by functional dysregulation of epigenetic proteins. Histone acetylation and deacetylation are important epigenetic reversible changes for gene expression, and altered expression is influential in cancer, neurological diseases, and immune disorders.1,2
Generally, acetylation of exposed ɛ-amino groups of lysine residues of histones H2A, H2B, H3, and H4 by histone acetyl transferase promotes gene transcription and noninteraction between DNA and nucleosome protein.3–5 In contrast, deacetylation of the same lysine residues by histone deacetylases (HDACs) enhances the interaction between DNA and nucleosome protein.6 In epigenetics, these reversible modifications of histones are important in regulating eukaryotic transcription and gene expression in the healthy state. In abnormal conditions, HDACs are overexpressed in human gastric, prostate, and colon cancers and in leukemia and lymphoma.7–12 For these reasons, the authors need to consider the balance between altered genetic and epigenetic regulation.
HDACs could be an attractive biomarker for cancer imaging.13 To date, 18 different HDACs have been characterized and classified into four groups (class I–IV) according to their structural diversity and catalytic functionality.14–16 HDAC inhibitors have been previously investigated as an antitumor agent to control cell death, differentiation, cell cycle arrest, DNA repair inhibition, upregulation or reactivation of silenced tumor suppressors, and the downregulation of growth factors, oxidative stress, autophagy, and angiogenesis.17 HDAC inhibitors, such as suberanilohydroximic acid (SAHA) and romidepsin (FK-228), have been developed and approved by the U.S. Food and Drug Administration (FDA) for treatment of cutaneous T cell lymphoma and other peripheral T cell lymphomas.18–21
Radiolabeled positron emission tomography (PET) imaging probes like SAHA have been used to synthesize 6-([18F]fluoro acetamide)-1-1hexanoicanilide ([18F]FAHA) for PET imaging of breast carcinoma in rats, human glioblastoma multiform, and baboon brain.22,23 However, the probe displayed a lack of the target binding affinity due to its low hydroxamate chelate activity and was rapidly metabolized in vivo. [18F]Fluorosuberoylanilide hydroxamic acid ([18F]SAHA) was evaluated in a murine ovarian cancer model,24,25 and [18F]fluoroethylsuberoylanilide hydroxamic acid ([18F]FESAHA) was evaluated concerning targeting of HDAC expression in LNCaP androgen-sensitive human prostate adenocarcinoma cell xenografts in mice.26 Click chemistry involving copper(I)-catalyzed Huisgen 1,3-dipolar cycloaddition is still one of the most exciting approaches to prepare targeted molecular imaging probes for PET by radionuclide labeling of small molecules, peptides, and proteins.27,28 A number of novel radiotracers have been reported through the use of click chemistry. So, the authors applied click chemistry to the synthesis of [18F]1 and evaluated its efficacy as a potential PET probe in labeling HDACs using a rat model of RR1022 sarcoma tumor.
Materials and Methods
Synthesis of methyl 8-oxo-8-(prop-2-yn-1-yl amino)octanoate (2)
Triethylamine (19.0 mL, 0.14 mol) and propargylamine (6.8 μL, 10.66 mmol) were dissolved in 10 mL dichloromethane (CH2Cl2) and stirred at 0°C in an ice bath. Methyl 8-chloro-8-oxooctanoate (1.3 mL, 7.10 mmol) was added to the reaction mixture. The reaction mixture was stirred at 0°C for 2 h and quenched with water (10 mL). The organic layer was collected, and the aqueous layer was extracted with CH2Cl2 (3 × 50 mL). The combined organic layer was washed with water and brine, dried over Na2SO4. The resulting solution was purified by semipreparative reverse phase (RP)-HPLC. HPLC condition: C18 column (YMC–Pack ODS–A, 5 μm, 250 × 10.0 mm) using water (A) and acetonitrile (B) under gradient conditions given below with a flow rate of 4 mL/min. The gradient conditions used are 5% (B) for 0–5 min; 5%–95% (B) from 5 to 20 min; 95% (B) for 20–25 min; with a dual-wavelength UV-vis detector followed by 210 nm and lyophilization yielded compound 2 as a yellow powder (15.1 mg, 71%). 1H-NMR (CDCl3, 400 MHz): δ 5.97 (s, 1H), 4.02–4.01 (q, J = 5 Hz, 2H), 3.63 (s, 3H), 2.29 (dt, J = 41.6 Hz, 7.6 Hz, 4H), 2.21 (t, J = 2.2 Hz, 1H), 1.65–1.55 (m, 4H), 1.32–1.29 (m, 4H); 13C-NMR (CDCl3, 100 MHz): δ 174.3, 172.8, 79.8, 71.6, 51.6, 36.3, 34.0, 29.2, 28.9, 28.8, 25.4, 24.8.
Synthesis of methyl 8-(((1-(2-hydroxyethyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-8-oxooctanoate (3)
Compound 2 (100 mg, 0.29 mmol) was added to a mixture of 3-butyn-1-ol (22.1 μL, 0.29 mmol), tert-butanol (2 mL), and water (1 mL). To this mixture was then added a water solution (1 mL) containing CuSO4·5H2O (26.8 mg, 0.08 mmol) and sodium ascorbate (40 mg, 0.20 mmol). The reaction mixture was then stirred at room temperature for 1 hour. Then CH2Cl2 was extracted from the mixture, washed with water, and dried over Na2SO4. The resulting solution was purified by semipreparative RP-HPLC. HPLC conditions: C18 column (YMC–Pack ODS–A, 5 μm, 250 × 10.0 mm) using water (A) and acetonitrile (B) under gradient conditions given below with a flow rate of 4 mL/min. The gradient conditions used are 5% (B) for 0–5 min; 5%–95% (B) from 5 to 20 min; 95% (B) for 20–25 min; with a dual-wavelength UV-vis detector followed by 210 nm and lyophilization yielded compound 3 as a yellow powder (81 mg, 76%). 1H-NMR (methanol-d4, 400 MHz): δ 7.87 (s, 1H), 5.50 (s, 1H), 4.48 (t, J = 5.2 Hz, 2H), 4.42 (s, 2H), 3.93 (t, J = 5.2 Hz, 2H), 3.65 (s, 3H), 3.35 (s, 1H), 2.33 (dt, J = 39.6 Hz, 7.6 Hz, 4H), 1.62–1.58 (m, 4H), 1.34–1.32 (m, 4H); 13C-NMR (methanol-d4, 100 MHz): δ 176.1, 175.9, 146.1, 124.9, `4.6, 53.9, 52.0, 36.8, 35.6, 34.7, 29.9, 29.8, 26.7, 25.8; high resolution mass spectrometry (HRMS) (electrospray ionization [ESI]): [M+H]+ calculated for C14H25N4O4 313.1876, found 313.1875.
Synthesis of methyl 8-(((1-(2-fluoroethyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-8-oxooctanoate (5)
Compound 3 (50 mg, 0.16 mmol) was dissolved in 10 mL CH2Cl2 and cooled with stirring to −40°C in an ice bath. At −40°C, (diethylamino)sulfur trifluoride (DAST; 77.5 mg, 1.59 mmol, 63.5 μL) was added dropwise, and the reaction mixture was warmed to room temperature over the course of 2 h. Then 50 mL of CH2Cl2 was added to the reaction solution, and the reaction mixture was extracted using 10 mL NaHCO3 (aq). The combined organic fractions were dried with Na2SO4, and the solvent was removed solvent in vacuo. The resulting mixture was purified by semipreparative RP-HPLC. HPLC condition: C18 column (YMC–Pack ODS–A, 5 μm, 250 × 10.0 mm) using water (A) and acetonitrile (B) under gradient conditions given below with a flow rate of 4 mL/min. The gradient conditions used are 5% (B) for 0–5 min; 5%–95% (B) from 5 to 20 min; 95% (B) for 20–25 min; with a dual-wavelength UV-vis detector followed by 210 nm and lyophilization yielded compound 5 as a white powder (23 mg, 60%). 1H-NMR (chloroform-d, 400 MHz): δ 7.66 (s, 1H), 6.24 (s, 1H), 4.86 (dt, J = 46.8 Hz, 4.6 Hz, 2H), 4.70 (dt, J = 26.8 Hz, 4.6 Hz, 2H), 4.52 (d, J = 5.2 Hz, 2H), 3.65 (s, 3H), 2.30 (dt, J = 38.4 Hz, 7.2 Hz, 4H), 1.64–1.58 (m, 4H), 1.33–1.29 (m, 4H); 13C-NMR (chloroform-d, 100 MHz): δ 174.3, 173.2, 123.4, 82.4, 80.7, 51.6, 50.8, 50.6, 36.5, 34.9, 34.1, 28.9, 25.5, 24.8; HRMS (ESI): [M+H]+ calculated for C14H24N4O3F 315.1832, found 315.1835.
Synthesis of N1-((1-(2-fluoroethyl)-1H-1,2,3-triazol-4-yl)methyl)-N8-hydroxyoctanediamide (1)
Compound 5 (0.4 g, 1.28 mmol) was suspended in a 6:1 (v/v) mixture of methanol and 50% hydroxylamine (aq) 14 mL. NaOH (aq) (1 N, 2 mL) was added to this suspension with stirring at room temperature. After 4 h, 2 mL of 1 N HCl was added to reaction mixture to neutralize the reaction. The resulting product precipitated. The resulting solution was purified by semipreparative RP-HPLC. HPLC condition: C18 column (YMC–Pack ODS–A, 5 μm, 250 × 10.0 mm) using water (A) and acetonitrile (B) under gradient conditions given below with a flow rate of 4 mL/min. The gradient conditions used are 5% (B) for 0–5 min; 5%–95% (B) from 5 to 20 min; 95% (B) for 20–25 min; with a dual-wavelength UV-vis detector followed by 210 nm and lyophilization yielded compound 1 as a yellow powder (255 mg, 52%). 1H-NMR (methanol-d4, 400 MHz): δ 7.88 (s, 1H), 4.90 (dt, J = 46.8 Hz, 4.6 Hz, 2H), 4.74 (dt, J = 26.8 Hz, 4.6 Hz, 2H), 4.25 (d, J = 5.2 Hz, 2H), 2.22 (dt, J = 38.4 Hz, 7.2 Hz, 4H), 1.67–1.54 (m, 4H), 1.33–1.31 (m, 4H); 13C-NMR (methanol-d4, 100 MHz): δ175.9, 172.7, 146.3, 124.7, 83.5, 81.8, 51.7, 51.5, 36.5, 35.3, 33.4, 29.6, 26.5, 26.3; HRMS (ESI): [M+H]+ calculated for C13H23N5O3F 316.1785, found 316.1786.
A 10 mL vial was charged with 30 mCi (1.11 GBq) [18F] fluoride (n.c.a), K2CO3 (3.8 mg) in water (200 μL), kryptofix 2.2.2 (16 mg), and 900 μL of CH3CN. The water was removed azeotropically using a heating block at 90°C under a flow of nitrogen. Azeotropic drying was repeated by the addition of anhydrous CH3CN (4°C, 1 mL). [18F]KF/Kryptofix complex (15.00 mCi (0.56 GBq)) was added in CH3CN (450 μL) containing 2-azidoethyl-4-toluenesulfonate (11 μL). The mixture was stirred at 90°C for 15 min. The resulting mixture was trapped into vial in ice bath by a stream of N2. The mixture added Cu(OAc)2 (1.2 mg, 0.007 mmol) and sodium ascorbate (1.8 mg, 0.006 mmol) and 2,6-lutidine 10 μL. The mixture was stirred at 60°C for 10 min and cooled down to room temperature. The resulting solution was purified by semipreparative RP-HPLC. HPLC condition: C18 column (YMC–Pack ODS–A, 5 μm, 250 × 10.0 mm) using water (A) and acetonitrile (B) under gradient conditions given below with a flow rate of 4 mL/min. The gradient conditions used are 5% (B) for 0–5 min; 5%–95% (B) from 5 to 20 min; 95% (B) for 20–25 min; with a dual-wavelength UV-vis detector followed by 210 nm and flow through gamma detector connected in series.
Biodistribution study of [18F]1 was carried out in normal ICR mice (male, 28–30 g, n = 5 per time point). Isoflurane-anesthetized mice were intravenously injected with 0.15 GBq of [18F]1 and sacrificed by cervical dislocation at 10, 30, and 60 min postinjection. Blood, heart, lung, liver, spleen, kidney, pancreas, stomach, small intestine, large intestine, bone, muscle, and brain were extracted, weighed, and counted on a gamma-counter. Results are expressed as percentages of injected dose per gram (%ID/g ± SD) of tissue with decay correction for normalized organ activity measurement to time of dose preparation.
Tumor xenograft model
All animal experiments were performed in compliance with the policies and procedures of the Institutional Animal Care and Use Committee for Animal Treatment, Chonbuk National University. A suspension of RR1022 sarcoma cells was injected subcutaneously [1 × 107 cells in 100 μL Matrigel+PBS (1:1, v/v)] into the right foreleg of female, 7-week-old Sprague-Dawley rats (Orient Bio, Seoul, Korea). The rats were examined by PET imaging when the tumor volume reached 2 cm in diameter (2 weeks after inoculation). During tumor growth, the tumor mass was maintained by intraperitoneal injection of cyclosporine (100 μL, 20 mg/mL). Tumor-to-blood ratio was measured using RR1022 sarcoma tumor-bearing rats.
Positron emission tomography/computed tomography
Micro-PET/CT images were acquired using a Gamma Medica FLEX™ X-PET®/X-O™ animal imaging instrument (X-SPECT system; Gamma Medica Ideas Inc., Northridge, CA), which combines a PET scanner and a multislice helical CT scanner. CT scans were performed using a CT scanner at 75 kVp and 0.25 mA. About 2 weeks after the tumor inoculation, RR1022 tumor-bearing rats were injected with 8.5 MBq (in 100 μL saline) of [18F]1 through the lateral tail vein using a catheter. Rats were anesthetized at 37°C using zoletil (Tiletamine Hydrochloride 145.5 mg+Zolazepam Hydrochloride 140.9 mg+diluent 5 mL+Rompun 10 mL [Xylazine Hydrochloride 23.32 mg, Sodium Chloride 3.00 mg]). PET scans were acquired for 30, 60, and 90 min postinjection. The PET/CT images were analyzed on a dedicated PMOD 3.7 workstation. Three-dimensional volumes of interest were drawn on the area of tumor and normal muscle regions, using the PMOD 3.7 software. Volumes of interest were drawn around the entire lesion with knowledge of the location of the tumor. In addition, similar regions of interest were drawn on normal muscle. The voxel value of the reconstructed image was converted to mean ± standard deviation.
Results and Discussion
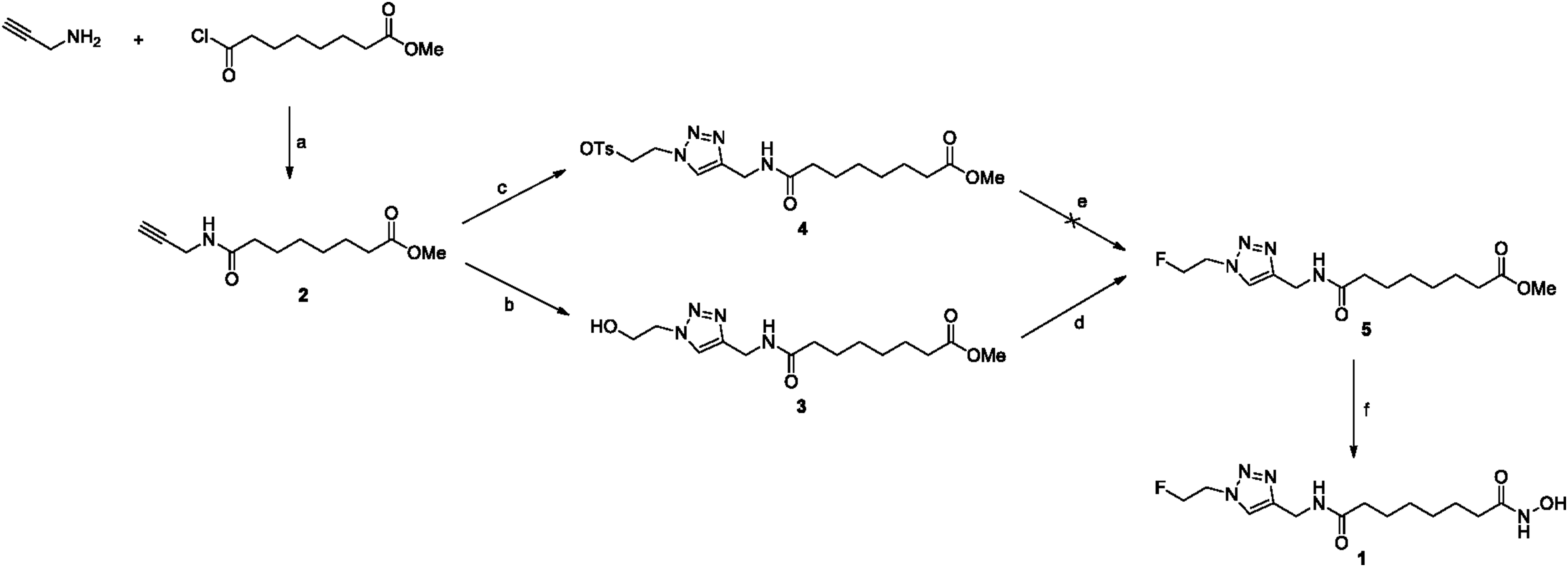
Introduction of a triazole moiety to the suberoyl hydroxamic acid for substitution of the aromatic ring of SAHA appeared to be a feasible approach using click chemistry. To identify radiochemical [18F]1, nonradiolabeled standard 1 was synthesized from methyl-8-chloro-8-oxooctanoate in four high-yielding steps (Fig. 1). Propargylamine was selected because it represents a terminal alkyne for click reaction with the azido compound. N-acylation between propargylamine and methyl-8-chloro-8-oxooctanoate was performed to generate the propargyl compound 2. Click reaction was performed with azidoethanol, in which the ethyl carbon chain was chosen as the radiolabeling tagging portion. Initial attempts to prepare the nonradiolabeled standard 1 from tosylate compound 4 using metal fluoride reagents, such as CsF and KF, and tetrabutylammonium fluoride (TBAF) for phase transfer method failed to give the desired product (Fig. 1). The fluorination of tosylate 4 with fluoride ion source under typical reaction conditions produced the eliminated alkene formed as a by-product. The same reaction in an aprotic solvent, such as tert-butanol (t-BuOH) and tert-amyl alcohol, yielded an eliminated alkene by-product that was analyzed by 1H-nuclear magnetic resonance (NMR).29,30
The authors considered fluorination of OH group compound 3 to avoid side product formation. The hydroxyl group of compound 3 was converted to fluorine using diethylaminosulfur trifluoride (DAST) as a fluorination reagent to yield the desired fluoro compound 5 (Fig. 1). Finally, N-hydrolysis reaction was performed with 50% NH2OH in NaOH to obtain the nonradiolabeled standard 1 in a yield of 52%. It was characterized by 1H-NMR and HR mass spectrometry (MS) m/z ES(+) (found:316.1786; calc.:316.1785) for purity and identity, respectively (Supplementary Figures S1–S7; Supplementary Data are available online at www.liebertpub.com/cbr).
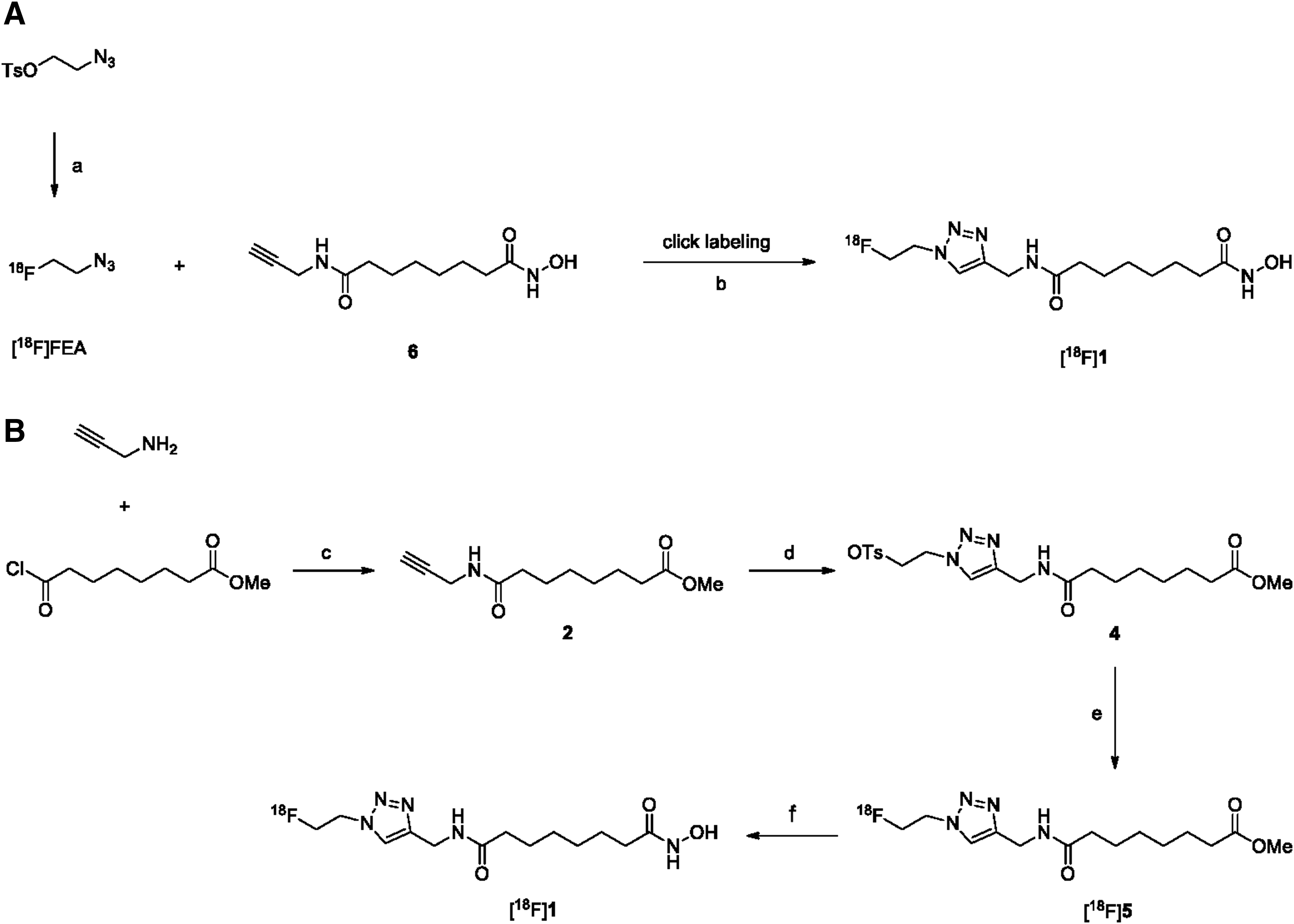
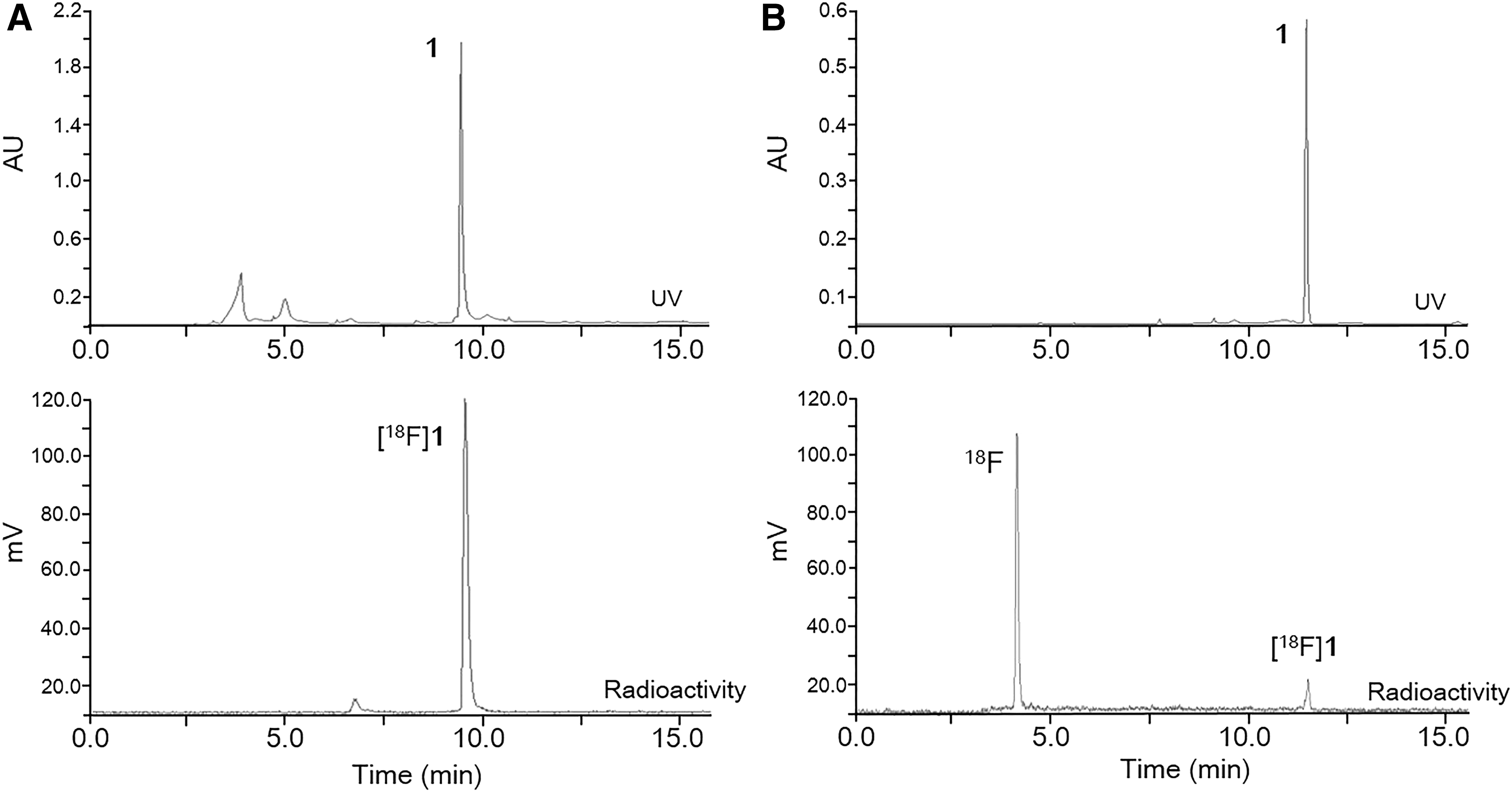
There are two primary approaches that have been reported for radiochemical synthesis of [18F]1 (Fig. 2). One approach includes conventional nucleophilic substitution of the triazole tosylate compound 4 with K[18F]F in the presence of a protic solvent, such as tert-butyl alcohol, and tert-amyl alcohol as a reaction medium (Fig. 2B). The approach results in low yield of product (less than 10%) (Fig. 3B), necessitating a subsequent N-hydrolysis reaction of the methyl ester moiety from [18F]5 using 50% NH2OH. The approach is time consuming. To do better, an alternative conjugation strategy based on click chemistry was used. Click chemistry involving the Cu(I)-catalyzed 1,3-cycloaddition of terminal alkynes with azides is a useful conjugation method (Fig. 2A). The approach is a simple, regioselective, and high-yielding reaction. Protection of other functional groups may not be required. In the click chemistry approach, [18F]fluoroethyl azide ([18F]FEA) was synthesized as a 18F-prosthetic synthon and distilled with acetonitrile into the reaction vial containing propargyl compound 6,31 CuSO4, and sodium ascorbate at room temperature. The radiochemical yield of [18F]FEA was 55.3% ± 4.0% (n = 6) based on radioactivity measurement from distillate to vial. The click reaction between [18F]FEA and terminal alkynes showed a good-to-excellent radiochemical yield of 31.2% ± 4.6% (n = 6) (Fig. 3A), and the total synthesis time, including high-performance liquid chromatography (HPLC) purification and reformulation, was 120–130 min. The desired product [18F]1 was identified by co-eluting with nonradiolabeled standard compound 1 on HPLC (Fig. 3). The radiochemical stability of [18F]1 was determined by incubation in normal saline and human serum at 37°C for 2 h with analysis by radio-thin layer chromatography (radio-TLC). Chemical stability was high (90%), indicating the relatively high in vitro stability of the radiotracer.
HPLC comparison with click reaction with [18F]FEA and terminal propargyl compound 6 (A) and nucleophilic substitution of tosylate compound 4 with K[18F]F (B) and HPLC profile of [18F]1 (bottom) and correlation with nonradiolabeled compound 1 in the UV (210 nm) chromatogram (top). HPLC condition (A) The gradient condition was used 5% CH3CN for 0 to 5 min; 5%–95% CH3CN from 5 to 20 min; 95% CH3CN for 20 to 25 min with NaI (Tl) detector (tR = 9.6 min). HPLC condition (B) The gradient condition was used 20% CH3CN for 0 to 15 min; 20% to 95% CH3CN from 15 to 30 min with NaI (Tl) detector (tR = 11.9 min). HPLC, high-performance liquid chromatography.
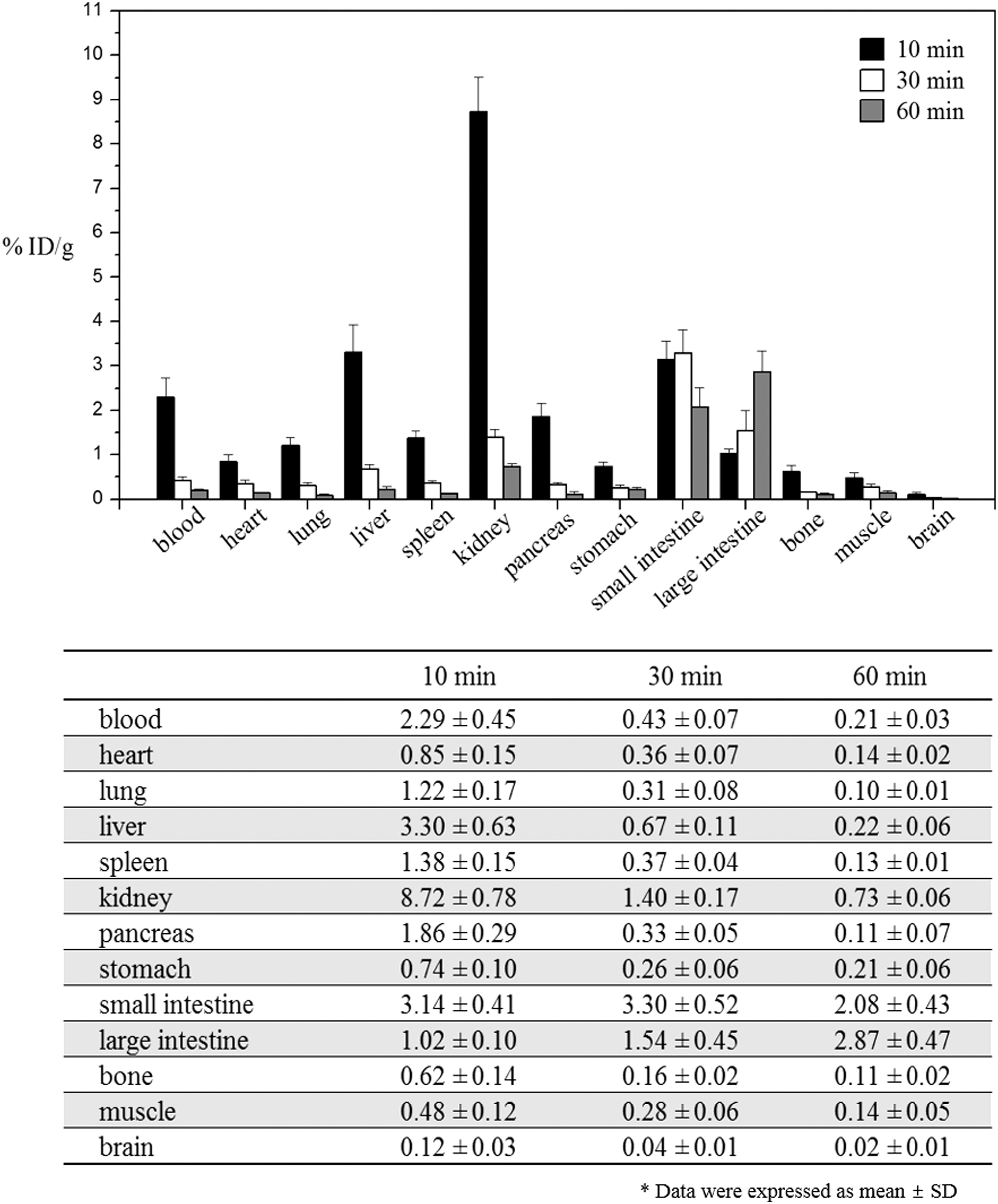
Biodistribution study of [18F]1 was performed with normal ICR mice. Mice were fasted for 14 h before administration of [18F]1 by intravenous injection and sacrificed at 10, 30, and 60 min postinjection. Subsequently, tissues were extracted, weighed, and counted on a gamma counter. High radioactivity was accumulated in kidney, liver, and blood and the radioactivity washed out rapidly with time from all tissues except for small and large intestine (Fig. 4). Kidney uptake (8.72 ± 0.78, 1.40 ± 0.17, and 0.73 ± 0.06%ID/g at 10, 30, and 60 min, respectively) decreased significantly, indicating fast renal clearance of the radiotracer. Small and large intestine radioactivity accumulation (small intestine: 3.14 ± 0.41, 3.30 ± 0.52, and 2.08 ± 0.43%ID/g, large intestine: 1.02 ± 0.11, 1.54 ± 0.45, and 2.87 ± 0.47%ID/g at 10, 30, and 60 min, respectively), however, did not decrease significantly because of gut uptake over the course of the study. Brain uptake (0.12 ± 0.03, 0.04 ± 0.01, and 0.02 ± 0.01%ID/g at 10, 30, and 60 min, respectively) was low, suggesting that [18F]1 could not penetrate the blood–brain barrier. No significant uptake was observed by bone, indicating that defluorination of [18F]1 was not regenerated by dehalogenase enzymes during the study. In case of [18F]FESAHA, bone uptake by defluorination of radiotracer was 9.6 ± 2.18, 11.1 ± 1.60, and 13.4 ± 0.60 at 30, 60, and 120 min, respectively.26
Biodistribution of [18F]1 in normal ICR mice (n = 5). Data are expressed as percentage of the injected dose per gram of tissue (% ID/g ± SD) after intravenous administration of [18F]1 at 10, 30, and 60 min postinjection.
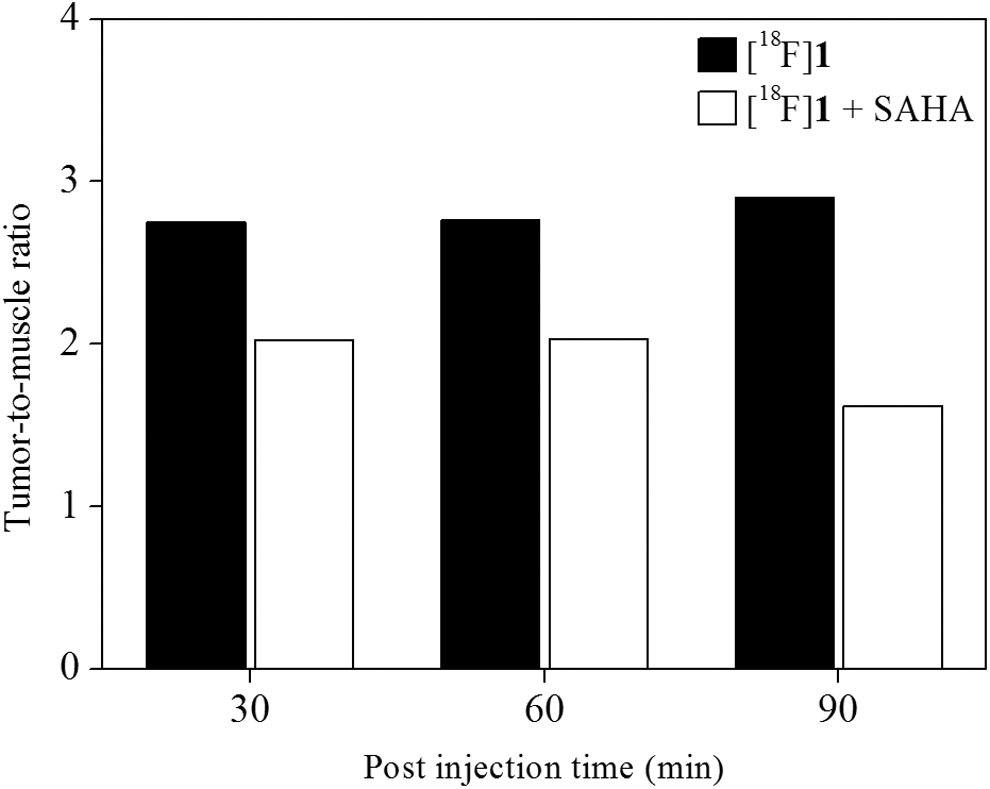
PET/computed tomography (CT) imaging following implantation of RR1022 sarcoma cells in rats was performed to assess radiotracer accumulation over time. This was done to determine optimal uptake and tumor-to-muscle ratio on the region of interest (Fig. 5). Before the addition of [18F]1, rats were fasted for 14 h. The PET acquisition time was 10 min and was done 30, 60, and 90 min postinjection and reconstructed by two-dimensional ordered-subset expectation maximization (OSEM2D). PET images clearly revealed high tumor uptake of [18F]1 during the entire image acquisition time (Fig. 6). The percentage injected dose in the tumor region was 1.09%ID, 0.74%ID, and 0.57%ID with 2.75, 2.76, and 2.90 tumor-to-muscle ratio at 30, 60, and 90 min, respectively, after radiotracer injection (Fig. 5). To subsequently determine specific tumor accumulation of [18F]1, the authors measured tumor-to-blood ratio using tumor rat model. As expected, tumor-to-blood ratio of [18F]1 (1.21 and 1.83 at 30 and 60 min, respectively) increased to 51% at 60 min from 30 min postinjection over the course of the study.
Uptake ratio of tumor-to-muscle of [18F]1 (black bar) and inhibition study with SAHA (20 mg/kg) at 30, 60, and 90 min (white bar).
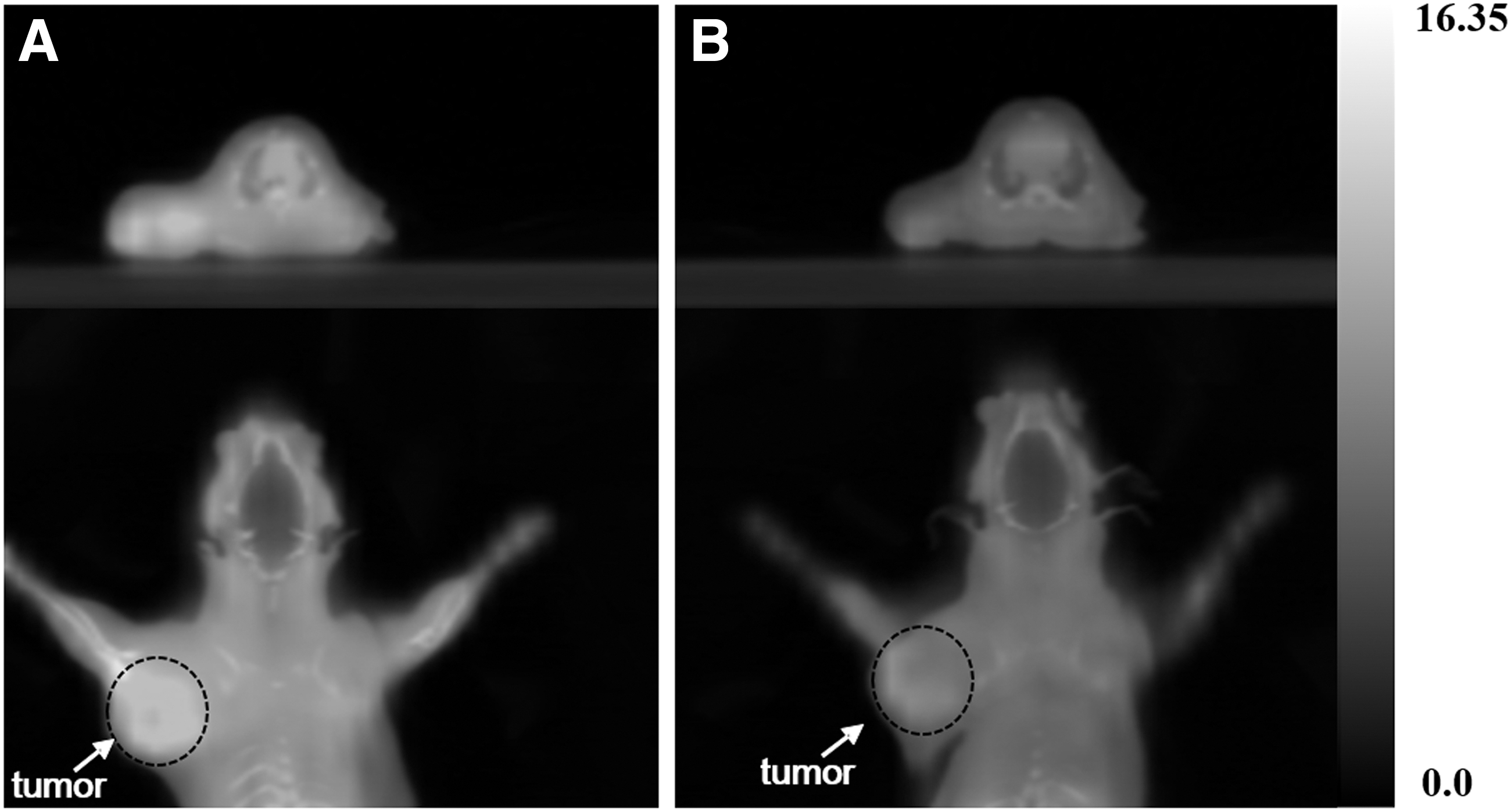
[18F]1 uptake in tumor inoculated rat model. (A) PET/CT images (top: transaxial and bottom: coronal) displaying RR1022 tumor rat intravenous injected with [18F]1 at 60 min postinjection and (B) inhibited PET/CT images with SAHA (inhibitor, 20 mg/kg) at 60 min postinjection. White arrow and dot mark the tumor region. PET/CT images were analyzed using PMOD 3.7 workstation. PET/CT, positron emission tomography/computed tomography.
These results demonstrated that the HDAC-rich tumor region was clearly visualized in PET imaging of rats injected with [18F]1. In addition, PET images revealed accumulation of radioactivity in the kidney and intestine, indicating the predominance of renal excretion, with high background activity noted in the gastrointestinal track. No significant defluoride from [18F]1 uptake at the bone was observed up to 90 min postinjection. To investigate the effective blocking of the tumor and normal organs, an inhibition study in tumor-bearing rats was carried out using SAHA, which was approved by the FDA as the first HDAC inhibitor.18 Before the addition of [18F]1, SAHA (20 mg/kg) was injected into the tumor-bearing rats, and its effects on the uptake of [18F]1 in the tumor were assessed. Representative PET images of RR1022 tumors after injection of [18F]1 in the presence of SAHA as an inhibitor are shown in Figure 6B. Tumor uptake was reduced to 0.55%ID, 0.28%ID, and 0.26%ID, and these results showed 49.5%, 62.2%, and 54.4% inhibition rate by SAHA at 30, 60, and 90 min, respectively (Fig. 7).
Percentage inject dose (%ID) of [18F]1 in tumor at 30, 60, and 90 min postinjection (black bar) and inhibition study using SAHA (gray bar).
Tumor-to-muscle ratio of inhibition experiment with SAHA was 2.02, 2.03, and 1.62 at 30, 60, and 90 min, respectively (Fig. 5). SAHA has high binding affinity to HDAC due to the presence of a metal binding domain that interacts with the active site, a linker domain that occupies the channel, and a surface recognition domain that interacts with residues. Radiotracer [18F]1 has a triazole moiety that, using the click reaction, substitutes for the aniline group for HDAC recognition. The result was the high inhibition rate (50%–62%), indicating that triazole moiety of [18F]1 avidly recognizes the active site of HDACs. According to the above results, [18F]1 may be superior to other HDAC PET imaging agents such as [18F]SAHA and [18F]FESAHA in terms of its high tumor targeting radioactivity fast clearance in blood and kidney and no defluorination of radiotracer.
Conclusion
[18F]FETSAHA ([18F]1) was successfully synthesized in high radiochemical yield using the click labeling method with [18F]FEA and terminal alkyne compound 6. PET evaluation of [18F]1 demonstrated marked accumulation of radioactivity in tumors and a high tumor-to-muscle ratio at all time points. [18F]1 is implicated as a potent radiotracer because of its specific uptake in tumors due to its high binding affinity to the HDAC receptor in a rat model.
Footnotes
Acknowledgments
This project was supported by Radiation Technology R&D program (2016M2A2A7A03912640) through the National Research Foundation of Korea funded by the Ministry of Science ICT & Future Planning, and Biomedical Research Institute, Chonbuk National University Hospital and Ministry of Science, ICT and Future Planning (MSIP) in Korean government, and Korea Industrial Technology Association (KOITA) as “A study on the program (R&D 4-4) to support investment-linked R&D for growth of companies.”
Disclosure Statement
No competing financial interests exist.
References
1.
WagnerJM, HackansonB, LübbertM, et al.Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics, 2010; 1:117.
2.
LiJ, LiG, XuW. Histone deacetylase inhibitors: An attractive strategy for cancer therapy. Curr Med Chem, 2013; 20:1858.
3.
ChakravarthyS, ParkY-J, ChodaparambilJ, et al.Structure and dynamic properties of nucleosome core particles. FEBS Lett, 2005; 579:895.
DokmanovicM, PerezG, XuW, et al.Histone deacetylase inhibitors selectively suppress expression of HDAC7. Mol Cancer Ther, 2007; 6:2525.
8.
ChoiJH, KwonHJ, YoonBI, et al.Expression profile of histone deacetylase 1 in gastric cancer tissues. Jpn J Cancer Res, 2001; 92:1300.
9.
HalkidouK, GaughanL, CookS, et al.Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate, 2004; 59:177.
10.
WilsonAJ, ByunD-S, PopovaN, et al.Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem, 2006; 281:13548.
11.
SongJ, NohJH, LeeJH, et al.Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS, 2005; 113:264.
12.
AhmadzadehA, KhodadiE, ShahjahaniM, et al.The role of HDACs as leukemia therapy targets using HDI. Int J Hematol Oncol Stem Cell Res, 2015; 9:203.
13.
StimsonL, La ThangueNB. Biomarkers for predicting clinical responses to HDAC inhibitors. Cancer Lett, 2009; 280:177.
14.
BlanderG, GuarenteL. The Sir2 family of protein deacetylases. Annu Rev Biochem, 2004; 73:417.
BhallaKN. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol, 2005; 23:3971.
17.
WestAC, JohnstoneRW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest, 2014; 124:30.
18.
MannBS, JohnsonJR, CohenMH, et al.FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist, 2007; 12:1247.
19.
MarksPA, BreslowR. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol, 2007; 25:84.
20.
Campas-MoyaC. Romidepsin for the treatment of cutaneous T-cell lymphoma. Drugs Today, 2009; 45:787.
21.
RichonVM, EmilianiS, VerdinE, et al.A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc Natl Acad Sci, 1998; 95:3003.
22.
ReidAE, HookerJ, ShumayE, et al.Evaluation of 6-([18F]fluoroacetamido)-1-hexanoicanilide for PET imaging of histone deacetylase in the baboon brain. Nucl Med Biol, 2009; 36:247.
23.
HookerJM, KimSW, AlexoffD, et al.Histone deacetylase inhibitor MS-275 exhibits poor brain penetration: Pharmacokinetic studies of [11C]MS-275 using positron emission tomography. ACS Chem Neurosci, 2009; 1:65.
24.
HendricksJA, KeliherEJ, MarinelliB, et al.In vivo PET-imaging of histone deacetylases by 18F-suberoylanilide hydroxamic acid (18F-SAHA). J Med Chem, 2011; 54:5576.
25.
SeoYJ, MuenchL, ReidA, et al.Radionuclide labeling and evaluation of candidate radioligands for PET imaging of histone deacetylase in the brain. Bioorg Med Chem Lett, 2013; 23:6700.
26.
ZeglisBM, PillarsettyN, DivilovV, et al.The synthesis and evaluation of N 1-(4-(2-[18F]-fluoroethyl) phenyl)-N 8-hydroxyoctanediamide ([18F]-FESAHA), A PET radiotracer designed for the delineation of histone deacetylase expression in cancer. Nucl Med Biol, 2011; 38:683.
27.
KimDH, ChoeYS, ChoiJY, et al.17-[4-(2-[18F] Fluoroethyl)-1H-1,2,3-triazol-1-yl]-6-thia-heptadecanoic acid: A potential radiotracer for the evaluation of myocardial fatty acid metabolism. Bioconjug Chem, 2009; 20:1139.
28.
GlaserM, ÅrstadE. “Click labeling” with 2-[18F]fluoroethylazide for positron emission tomography. Bioconjug Chem, 2007; 18:989.
29.
KimDW, JeongHJ, LimST, et al.Facile nucleophilic fluorination reactions using tert-alcohols as a reaction medium: Significantly enhanced reactivity of alkali metal fluorides and improved selectivity. J Org Chem, 2008; 73:957.
30.
KimDH, ChoeYS, KimBT. Evaluation of 4-[18F]fluoro-1-butyne as a radiolabeled synthon for click chemistry with azido compounds. Appl Radiat Isot, 2010; 68:329.
31.
SmithG, GlaserM, PerumalM, et al.Design, synthesis, and biological characterization of a caspase 3/7 selective isatin labeled with 2-[18F]fluoroethylazide. J Med Chem, 2008; 51:8057.
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.