
Abstract
Secreted inflammatory cytokines are considered as critical mediators in the tumor microenvironment. Cancer cells play a significant role in the differentiation of effector T cells to exhausted T cells through mediator production. Effector T cells that undergo tumor microenvironment become terminally differentiated into exhausted T cells. These changes create an opportunity for tumorigenesis and invasion of cancer cells. Despite having some characteristics of effector T cells, the exhausted T cells lose their antitumor properties. In this article, the phenotypes and function of exhausted T cells were reviewed and the expression pattern of inflammatory cytokines in tumor tissues and peripheral blood of cancer patients were described. Additionally, the effects of inflammatory cytokines on intracellular factors and signals of T lymphocyte cells were explained. In conclusion, the authors referred to probable approaches that could be used to improve the antitumor activity of T cells and intervention into T cell exhaustion.
Introduction
The immune system recognizes cancerous cells by screening the tumor antigen during malignant progression. By producing cytokines, the cells of the immune system have the ability to arrest division and migration of cancer cells. 1 Cancer cells commonly change the cytokine profile so that the innate and adaptive immune cells are differentiated in the tumor microenvironment. 2,3 Cytotoxic T lymphocytes (CTL), which are exposed to the microenvironment in tumor tissues and peripheral blood, lose their antitumor activity and express inhibitor marker on their surface. In fact, these lymphocytes are transformed to exhausted T cells with different features in phenotype and performance. 2
Investigations have shown that the exhausted T cells give rise to resistance against chemotherapy and develop angiogenesis by interleukin (IL)-8 and IL-6 production. 4,5 The limitations in specific antigen diagnosis and T cell activation have led to the limitation of using novel immunotherapy techniques, such as chimeric antigen receptor (CAR) and T cell receptor (TCR) transgenic T cells. 6,7 Recently, some immune checkpoint inhibitors, such as ipilimumab, act against inhibitor markers and have replaced chemotherapy and radiotherapy in melanoma and some sorts of leukemia. Although these antibodies seem to be effective drugs to activate exhausted T cells, they create systemic complications, such as diarrhea in the gastrointestinal tract, rash, pancreatitis, and autoimmune diseases. 8,9
Further investigations on tumor microenvironments in different kinds of cancer can improve insights on immune system change in this condition. In this article, the authors reviewed the phenotypes and function of exhausted T cells and described the expression pattern of inflammatory cytokines in tumor tissues and peripheral blood of cancer patients. The effects of inflammatory cytokines on intracellular factors and signals are identified to clarify the inactivation state. Likewise, exhausted T cells in some kinds of chronic inflammatory diseases, which have similar immunological characters, were considered.
The Exhausted T Cells and Their Markers
Exhausted T cells are capable of eliminating cancer cells; however, they enter an inactive state due to their presence in the microenvironment in various infections, autoimmune diseases, and cancer. These lymphocytes receive combined signals after raised inhibitory markers, including PD-1 (programmed cell death-1), Tim-3 (T cell immunoglobulin mucin-3), LAG-3 (lymphocyte activation gene-3), and TIGIT (T immune receptor with Ig and ITIM domain). However, the loss of activator markers, such as (CD127)IL-7R, CD28, CD27, and IL-15R extensively decrease the function of exhausted T cells in response to external signals. Exhausted T cells are not able to mobilize and release granules, including IFN-γ and granzyme β that have dramatically been assembled in the lymphocytes. 10,11 The differentiation and the most significant features of exhausted T cells are shown in Figure 1.
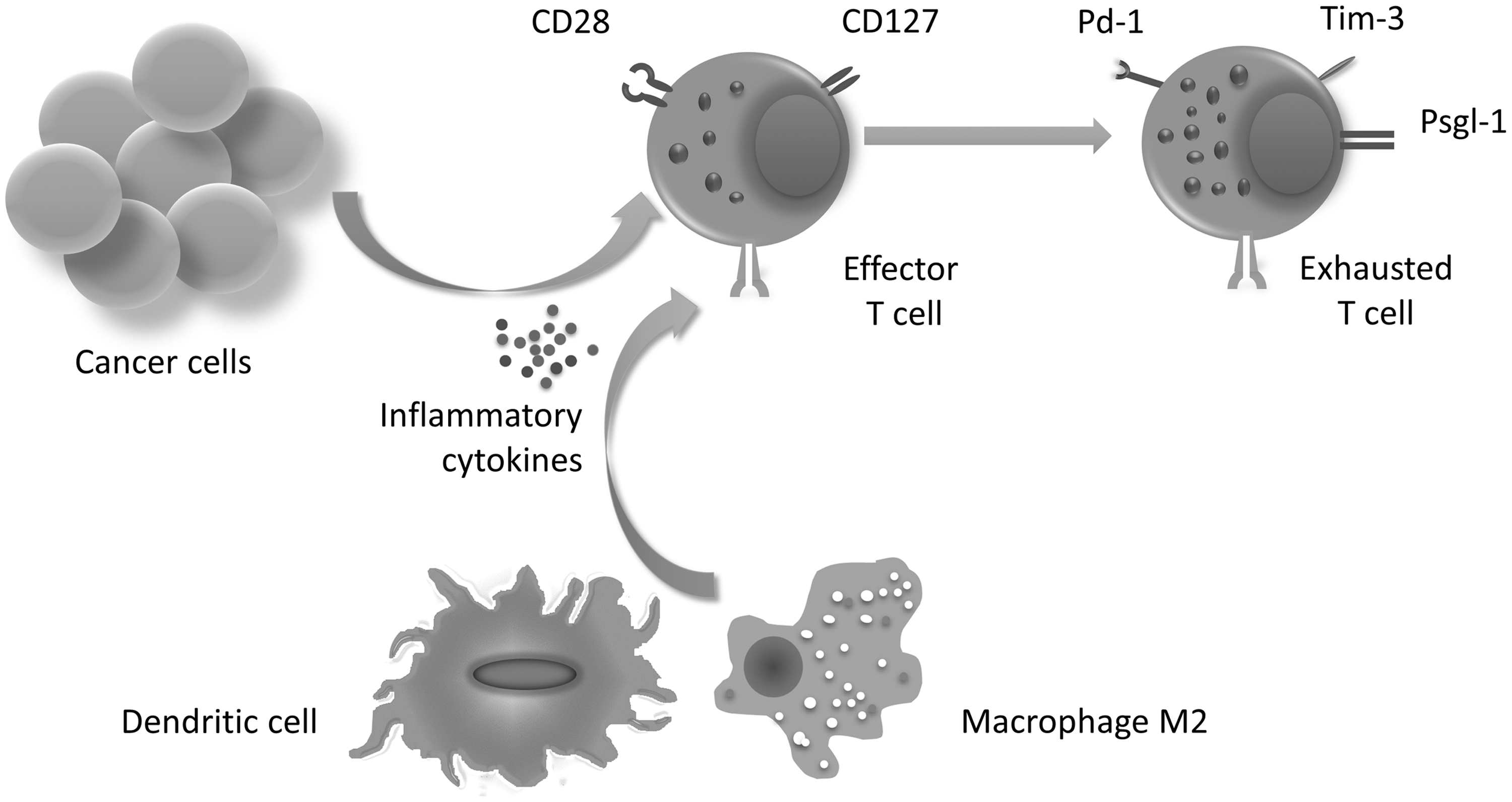
Tumor-derived inflammatory cytokines induce exhausted T cells. Cytokines produced by innate immune cell promote this differentiation. Attributed conditions lead to increased inhibitory markers and granules in exhausted T cells.
Oxidative phosphorylation, which is the central pathway for effector T cells to receive energy, is modulated in exhausted T cells. Alternatively, glut1 is significantly expressed in the exhausted T cells leading to elevated glycolysis pathway. 12,13 T cell exhaustion enriched in the tumor microenvironment are termed “Tumor-induced senescent T,” because they are similar to lymphocytes in older people. Senescent T lymphocytes face oxidative stress and inflammation conditions created by M2 macrophages and dendritic cells. These cells usually induce angiogenesis factors and have short length of telomeres and blunt cell cycle. 3,14
Programmed cell death-1
Surface cell marker PD-1 (CD279) is a member of the immunoglobulin CD28 superfamily, which have the potential to regulate the function of effector T cells. PD-1 intracellular domain transfers inhibitory signals that affect cell functions. PD-1 binds to PD-1L (B7-H1) found on cancer cells and antigen-presenting cells and PD-2L (B7-H2) on cancer cells, respectively. This immunoreceptor contains tyrosine-based inhibitory motif (ITIM) that recruits Src homology region two domain-containing phosphatase-1 (SHP-1), SHP-2, and SH2-domain-containing phosphatidylinositol 5-phosphatase. Phosphatase signals are then created when the immunoreceptor engages with a ligand. This signal represses the function of serine–threonine kinase Akt and phosphoinositide 3-kinase, which sustains retinoblastoma factor dephosphorylation. On the other hand, mammalian cyclin-dependent kinase inhibitor remains stable against the effects of UbQ ligase-SCF, and consequently, the cell cycle of T cells is stopped. 15,16 The downstream signal of PD-1 motivates basic leucine zipper transcription factor ATF-like (BATF) by which PD-1 expression is induced through a positive feedback. 17 In T cell exhaustion, pd-1 gene promoter (pdcd1) remains demethylated in contrast to the other T lymphocytes. This state is coupled with high amounts of transcription factors, such as NFAT, STAT3, BLIMP, and EOMES, which directly control different areas of the pdcd1 to be active. 9,18 Conversely, transcription factor T-bet is downregulated in patients suffering from cancer. T-bet further modulates PD-1 expression. As a result, all these conditions help to promote PD-1 expression. 19,20 Investigations on colorectal cancer in experimental mice treated by MAPK inhibitor and injected anti-PD-1L antibodies showed that the T-bet of CD8+ T cells increased while tumor size decreased. Although MAPK is very important for IL-2 production and cell activation in naive T cells, a continuous exposure to antigen by lymphocyte's TCR results in encoding nur77 gene and CTL apoptosis. 21 A fraction of NY-ESO-1-specific CD8+ T cells in patients with advanced melanoma expressed PD-1, Tim-3, and LAG-3 simultaneously. It offered the possibility that NY-ESO-1-specific T cells upon prolonged antigen stimulation, along with the interaction of PD-1 and ligand, resulted in upregulation of the inhibitor marker expression. 22
T cell immunoglobulin mucin-3
This marker can activate intracellular attenuation of T cells in response to galectin-9. This connection triggers inhibitor signals in addition to releasing an influx of Ca2+ into the lymphocytes, and thus induces apoptosis. The coexpression of PD-1 and Tim-3 has been reported in some kinds of cancers and virus infections. 23 Simultaneous injection of anti-PD-1 and anti-Tim-3 antibody in a mice model of breast, colorectal, and melanoma tumor produced more of the IFN-γ, which eventually controlled tumor growth. The prescription of anti-PD-1 alone for lung cancer patients resulted in increasing Tim-3 expression. 24,25 Tim-3 intracellular domain links to HLA-Bat3 (B-associated transcript 3) in playing a pivotal role in the proliferation and production of inflammatory cytokines. 26 Tim-3 intracellular signal in dendritic cells arrests HMGB1 pathway by which DNA antigens are recognized through TLR-4. Therefore, the efforts of the innate immune system against a cancerous mass would be in vain. 27
P-selectin glycoprotein ligand 1
P-selectin glycoprotein ligand 1 (PSGL-1) is a mucin-like glycoprotein observed on the surface of myeloid, lymphocyte, and endothelial cells. Psgl-1 binds to lymph node homing molecules, such as L-selectin. Conversely, it can also join to P and E-selectin related to cadherin, which facilitates lymphocyte movement into inflammatory regions. 28 Likewise, CCL-19 and CCL-21 chemokines are capable of interacting with Psgl-1 on the memory and naive T cells to trigger inhibitor signal into cells. Psgl-1 correlates PD-1 expression on the CD8+ T cells in melanoma mice and LCMV-C13-infected mice. Eliminating psgl-1 gene in CD8+ T cells leads to the elevation of T-bet, IL-2/7/15R, and reduction of PD-1 and Tim-3. 29,30 ERM (EZRIN/RADIXN/MOESIN) covers other markers of immunoreceptor tyrosine-based activation motif (ITAM) and interacts with the Psgl-1 intracellular domain. Accordingly, ITAM domain is not detected by Syk in the presence of Psgl-1 signals. ERM, along with Csk-binding protein (Cbp), activates c-Src associated with EBP50 protein. c-Src prohibits the formation of a mature immunological synapse to respond to phosphorylating lymphocyte-specific protein tyrosine kinase (Lck) in T cells. 31,32
CD127
This marker is the α chain of the IL-7Rα that gives rise to antiapoptotic molecules in the lymphocytes to sustain protective immunity. CD127 may undergo endocytosis when a signal transducer or activator of transcription 5 (STAT5) continually remains active through positive feedback; however, the lack of CD127 in the T cell demonstrates a significant impaired IL-2 secretion. 33,34 Investigations on peripheral blood of patients with breast cancer showed that coexpression of CD127 and CD28 was less frequent among PD-1+ T cells, particularly in a fraction of HER2-Neu+-specific T cells. T cells were probably under the effect of PD-1 signals. 35 The simultaneous use of dendritic cell therapy and anti-PD-1L antibody in mice with breast cancer led to restoration of T cell activity and CD127 marker production. Indeed, T cell exhaustion produced effector function that had previously been lost during the disease. 36
CD28
CD28 is a human T-cell-specific homodimer surface protein, which engages with its ligand (B7) to provide an essential costimulatory signal for naive T cells. The irreversible loss of CD28 is found on T cells in patients with advanced breast and lung cancers. 37,38 CD28 activity prompted cell cycle in T cells containing PD-1+ CD28− markers. These attributed features highlight the crucial role of eliminating CD28 in transition effector state into exhaustion. 37,39
Inflammatory Cytokines in Cancer
The performance of cancer and innate immune cells extensively has the potential to engender an immunosuppressive state. They produce mediators and inflammatory chemokines either recruiting immune cells toward a tumor, or inducing cancer cell migration to other limbs. 40,41 Comprehensive profiling of proinflammatory cytokines, including IL-1, IL-6, and tumor necrosis factor-α (TNF-α) is expressed in the tumor microenvironment. Proinflammatory cytokines highly induce angiogenesis, metastasis, and inflammatory chemokine in addition to directly affecting cancer cells. According to 177 studies recently carried out, IL-6 boosts malignance among leukemia and multiple myelomas. 42 Likewise, IL-6 variants are associated with susceptibility to afflict colorectal cancer. 43 The existence of large amounts of angiogenesis factors, such as CD15, CD68, and CD31, has been documented in tumor tissue of patients suffering from carcinoma and adenocarcinoma cancers as a result of increasing IL-6 expression in blood and tumor tissue. 44 TNF-α is of much importance in terms of generating inflammatory conditions. In spite of releasing low dose of TNF-α by means of a wide range of cancer cells, this cytokine has the potential to stimulate dendritic cells and macrophages. Inflammatory conditions are chronically sustained in breast, lung, colorectal cancer, and myeloid lymphoid malignancies due to the presence of large amounts of TNF-α. 45,46 Increased levels of IL-1 that activates NF-κB signaling pathways are observed in patients with metastatic melanoma, colorectal cancer, and nonsmall cell lung cancer. Leptin in obese patients with breast cancer contributes to producing IL-1, IL-8, and finally angiogenesis factors, such as vascular endothelial growth factor (VEGF). 47,48 Similarly, the abundance of prostaglandin E1/E2 (PGE1/PGE2) and VEGF factors is linked to the development of TH17 subset in Hodgkin and non-Hodgkin lymphoma, hepatocellular carcinoma to recruit macrophages and neutrophil into tumor tissue, as well as IL-6 production. 49,50 On the other hand, IL-21 of TH17 activates JAK-STAT1/3 pathway in adult T cell leukemia/lymphoma and multiple myeloma so that replicative potential and proliferation are highly promoted in cancer cells. 51
Signal Inducing T Cell Exhaustion by Inflammatory Cytokines
As IL-21 and IL-6 engage their receptors on T cells in the tumor microenvironment, the JAK-STAT3 signal has two downstream consequences. At first, the activated STAT3 plays a transcriptional factor role for pd-1 gene. Second, this signal activates BATF factor-binding c-jun, which is a part of activating protein-1 (AP-1) structure. In other words, BATF replaces foc factor eventually causing AP-1 not to be formed. As a result, NFAT factor expresses pd-1 gene alone, instead of interaction with AP-1 to produce IL-2 and IL-4. 52,53 The balance between BATF factor and EGR2/3 factor results in IL-2 production, whereas the decreased level of EGR2/3 helps produce IL-6 and IL-21. The high expression of BATF is accompanied with BCL-6, c-MAF, SAP-1 (SRF accessory protein-1), and PD-1 level in angioimmunoblastic T cell lymphoma disease. 54,55 BATF and IRF-4 (interferon regulatory factor 4) simultaneously encode BLIMP, which is one of the transcription factors of the pd-1 gene; however, the PD-1 intracellular domain motivates BLIMP through an unknown way. BLIMP is necessary for naive T cells to become terminally differentiated into effector T cells, and prevent deviation into memory T cells. The coexpression of PD-1 and BLIMP is observed in chronic viral infection. 56 Functionally, the combination of BATF and c-Maf may provide positive feedback to regenerate JAK-STAT3 active, which can act as IL-6 and IL-21 transcription factors. 57 Signals derived from tumor suppressor and inflammatory cytokine can offer the possibility to be integrated and switch T cell fate. For instance, TGF-β induces FOXP3 that can combine with STAT3 to form STAT3-FOXP3, an active heterodimer transcription factor, repressing CD127 expression in T cells. 58,59 Figure 2 illustrates different signals by which T cells differentiate into T cell exhaustion.

A complex set of cytokines and ligands involved in T cell exhaustion induction. Interaction among transcription factors, such as FOXP3, STAT3, and BATF, increases the inhibitory markers. In contrast, CD127 is downregulated in T cell exhaustion.
T cell exhaustion is noticeably capable of maintenance and adaptation in tumor microenvironment, whereas the cell cycle and proliferation become arrested. T cell exhaustion manages to overcome apoptosis process to guarantee their existence. Indeed, STAT3 further modulates BCL-2 family member BAD and Fas-ligand while upregulating bcl-2 antiapoptotic and ox-40. Studies on patients with breast cancer display a high amount of FAS and FAS-L in early stages rather than advanced ones. However, FAS is widely found on tumor-infiltrating lymphocytes. 60 As hypoxic stresses are established in the tumor microenvironment, tumor-infiltrating lymphocytes can make intracellular attenuation to mediate mechanistic target of rapamycin (mTOR) pathway. mTOR stimulates several factors, including HIF-a, FOXO3, and CITED-CBP in the order so that apoptosis is prevented. 61 FOXO1 and FOXO3 are induced in response to inflammatory cytokine retarding IL-2 release and cell cycle. In addition, FOXO1 regulates the transcriptional activity of the pd-1 gene. 18,62 Il-21 triggers β-catenin pathway through tcf-7 and lef1 motivation, finally leading to reprogramming of T cell into stem memory T cell. 63,64 The controversial issue is whether IL-21 has an antitumor activity or not. There was an attitude that the cooperation of IL-21 with IL-7, IL-15, and IL-2 restored natural killer cell (NK) and CTL to promote immune attack against a tumor. It can be mentioned that not only does T cell exhaustion loses CD127, IL-2R, and IL-15R but also the effect of IL-21 alone differentiates T cell into T cell exhaustion. 65 Eliminating il-21 and il-21r gene in mice with a tumor or chronic viral infection creates a subtype of T cells in which KLRG-1 (killer cell lectin-like receptor G1) is overexpressed and the capacity of producing IL-2 is exterminated. 66,67 The transcription of cd28 gene is reduced in T cell exhaustion as a result of being exposed to TNF-α. In this respect, peripheral blood mononuclear cells (PBMCs) were cultured with Epstein–Barr virus (EBV)-infected lymphoblast while the blocking TNF-α has been treated. Assessments revealed increasing transcription of cd28 and caspase3 genes in CD28+ T subset. 68,69 Treating tumor-infiltrating lymphocytes in breast cancer with IL-2 gave rise to T cell division and IFN-γ production over a long period; however, the expression level of PD-1 Tim-3 remained unchanged. Likewise, making use of IL-15 had the same effects on T cell in short time. 70 The phenotype of tumor-infiltrating lymphocytes differentiates more into exhausted T cells simultaneously to progress in lung cancer stages. Consistent with this study, INF-γ, T-bet, and perforin1 reduce in the T cell to such an extent that cancer cells settle in safe conditions. The lack of INF-γ in the tumor microenvironment has a wide range of consequences. On the one hand, phosphorylation of STAT1a, a significant transcription factor for Th1 function, is further modulated. On the other hand, the dephosphorylation of STAT1a in macrophage augments colony-stimulating factor-1 (CSF-1) level. CSF-1 urges macrophage M2 to migrate into tumor tissue. 20,71 Based on another research, the high level of EGF in macrophage and cancer cells is produced in situations where CSF-1 and EGF help encode each other. So, the conditions are provided to start cancer cell metastasis. 72 Simulating tumor microenvironment indicates the relationship between innate and adaptive immune cells releasing inflammatory cytokine and angiogenesis factors, such as IL-8, MMP, and VEGF. Tim-3 and CD40L on T lymphocytes bind to galectin-9 and CD40, respectively, on monocytes and macrophages. 3 In spite of efforts made to restore T cell exhaustion into effector T cells, a mechanism has been proposed for reprogramming T cell exhaustion into central memory T cells. These cells contain antigen receptor, specifically capable of gendering effector T cell. Umbilical cord blood transplantation (UCBT) has been prone to delayed immunological reconstitution and late memory T cell skewing. UCBT treated with PGE2 displays high amounts of IL-7R and IL-2R due to deactivated GSK-3. β-Catenin motivates differentiation into stem cell memory T cell pathway through tcf-7. 73,74
T Cell Exhaustion in Inflammatory Diseases
According to phenotypes and functions attributed to T cell in patients with inflammatory disease, indications of exhausted T cell have been documented in some cases. T cell exhaustion with characteristics, including inflammatory cytokine production, resistance to apoptosis, reaction with self-antigen and alteration of polyclonal to be oligoclonal, has represented the predominant fraction of T cells. Incidentally, T cell exhaustion potentially resists against corticosteroid drug. 75,76 Studies on humans indicate that the T cells accumulate TNF, IFN-γ, IL-6, and IL-21 in chronic inflammatory diseases, such as Crohn's, ulcerative colitis, and rheumatoid arthritis. CD28 null T cells are enriched in these kinds of patients, particularly in advanced phases. 77 –80 In patients chronically infected with HCV, HBV, and HIV, the increased level of PD-1 and Tim-3, and irreversible loss of CD28, CD27, and CD127 are maintained on T cells, which seems to impair immune responses against viral infection. 81 –83 The role of T cell exhaustion in releasing inflammatory cytokine has been prominent in a variety of deleterious clinical outcomes in Multiple Sclerosis (MS). Findings clarify insight into CD4+ CD28 null T cells, which tend to implant in cerebrospinal fluid mediated by CX3CR1 expression and IFN-γ production. 84,85 Patients afflicted by HLH usually have trouble secreting granules from NK and T cells. Their T cell exhaustion bears granules, including TNF, IL-1/6, and INF-γ in addition to Tim-3 and PD-1 on the surfaces. In this respect, experimental animal studies carried out on Stx11 knockout mice show that mice have a deficiency in granule release, and inhibitory markers are found on their T cells. Viral infection causes them to raise inflammatory cytokine production. However, existing Tim-3 and PD-1 on T cell control the immune reaction. In fact, inhibitory markers help Stx11 knockout mice to survive. 86 Investigations on children with Helicobacter pylori infection correlated with the gastric inflammation and elevated proportion of cd8+ cd28− cd27− T cell. Other studies confirm that biopsies taken from the stomach of subjects infected with H. pylori display a high level of IL-6 and IL-21, chronic phase in particular. 87 –89
Conclusions
The production of inflammatory cytokines by cancerous cells is of much significance to change the immunological state of the tumor microenvironment. Studies illustrate that the inflammatory cytokines respond directly to create cell exhaustion through multiple pathways. According to the pathways discussed in this study, inhibitory signals can attenuate antitumor activity of T cells through inhibitory markers. In the process of T cell exhaustion, tumor microenvironments correlate with inflammatory cytokines. With regard to intracellular signals, effects of inflammatory cytokines, such as IL-1 and IL-6, on T cells were discussed. The increased portion of the T cell exhaustion in the inflammatory bowel disease, chronically infected, and autoimmune diseases compared with those of healthy people showed similarity in the immune system.
More efforts should be made to reprogram exhausted T cell into the early state or even therapeutic interventions into exhausted T cell. Anti-inflammation therapy is able to change immune system function in inflammatory conditions. Likewise, targeting factors, which are related to signals of inflammatory cytokines, can improve CAR-T cell and TCR transgenic T cell therapy. In addition, further studies are needed to clarify the role of IL-21 and its consequences on T cells. In sum, understanding the features of exhausted T cells can increase ones knowledge of cancer and outcomes in the immune system, which also occurs in chronic inflammatory diseases. It may provide an opportunity to improve existing treatment pathways.
Footnotes
Acknowledgments
The authors are grateful to the staff of Students Research Committee, Shahrekord University of Medical Sciences, Shahrekord, Iran. This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Disclosure Statement
No competing financial interests exist.