
Abstract
Background:
Glioma is considered one of the most prevalent and lethal brain tumors. Glioblastoma (GBM) is a main subtype of glioma. Long non-coding RNAs (lncRNAs) are identified as a new class of biomarkers and therapeutic targets for treatment of GBM.
Objective:
In the present study, we focused on exploring the function and potential mechanistic regulation of lncRNA small nucleolar RNA host gene 5 (SNHG5) in GBM.
Methods:
Gene expression was determined by qRT-PCR or western blot, as appropriate. CCK-8 and EdU assays, flow cytometry analysis and caspase 3 activity assay were conducted to evaluate several cellular processes in GBM cells. The relationship between YY1 and SNHG5 was assessed via ChIP and luciferase reporter assays.
Results:
SNHG5 was highly expressed in GBM. Loss- and gain-of-function assays revealed that SNHG5 promoted GBM cell proliferation and inhibited cell apoptosis in GBM. Mechanism experiments proved Yin Yang 1 (YY1) as transcriptional activator of SNHG5 in GBM. More importantly, we found that SNHG5 played the oncogenic role in GBM by activating p38/MAPK signaling pathway.
Conclusion:
YY1-induced SNHG5 promoted the cell proliferation in GBM via p38/MAPK signaling pathway. The findings expanded our understanding of SNHG5 as an oncogene in GBM.
Introduction
Among all malignant brain tumors, glioma is one of the most common forms, accounting for 70% of malignant primary brain tumors and 30% of central nervous system tumors. 1,2 As a main subtype of glioma, glioblastoma (GBM) is characterized by high incidence and high mortality. 3 Despite the advances in treatments for GBM, including the surgery combined with chemotherapy and radiotherapy, GBM remains incurable with poor prognosis. The treatment efficacy of GBM is hampered by strong cell proliferative ability and inherent resistance to chemotherapy and radiotherapy. 4 Hence, it is vital to explore novel biomarkers and therapeutic targets for patients with GBM.
Long noncoding RNAs (lncRNAs) are a type of transcripts longer than 200 nucleotides without protein-coding potential. 5 Dysregulation of lncRNAs is reported widely to have an impact on cellular processes of tumors, particularly in proliferation and apoptosis. 6 –9 Mounting evidences suggest that aberrant expressions of lncRNAs are associated with biological behaviors of GBM. 10 –12 LncRNA small nucleolar RNA host gene 5 (SNHG5) attracts increasing attention of researchers owing to its oncogenic role in cancers such as colorectal cancer, osteosarcoma, gastric cancer, and other malignancies. 13 –17 Nevertheless, the biofunction and underlying regulatory mechanisms of SNHG5 in GBM development remain ill defined. Based on the previous reports, the alteration of cell proliferation ability is of great significance for the initiation and development of GBM. 18,19 Thus, they intended to investigate whether SNHG5 influenced GBM cell proliferation and apoptosis. With that, they designed and carried out a sequence of loss and gain-function assays in GBM cells.
Accumulating studies reveal that transcription factors contribute to lncRNA transcription, thereby enhancing the expression of lncRNAs in a variety of tumors. 20 –23 LncRNA could exert their functional involvement in modulating cancerous cellular processes. 24 –26 On the basis of this notion, they designed to further investigate the upstream molecular mechanism related to the aberrant expression of SNHG5 at the transcriptional level in GBM.
The p38/MAPK signaling pathway is a highly conserved pathway that mainly modulates cellular activity related to cancerous development, including proliferation, differentiation, and apoptosis. 27,28 Importantly, previous study showed that p38/MAPK signaling pathway was involved in lncRNAs mediated progression of GBM. 29 However, the correlation between SNHG5 and p38/MAPK pathway is yet to be explored. Hence, mechanism assays were performed to evaluate the role of p38/MAPK pathway in SNHG5-mediated regulation in GBM.
Materials and Methods
Cell culture
The normal brain glial cell line HEB and three human GBM cell lines (U251, U87 and LN229) were purchased from American Type Culture Collection (ATCC, Manassas, VA). The four cell lines were routinely cultured in RPMI-1640 medium (Invitrogen, Carlsbad, CA). Ten percent fetal bovine serum (FBS; Gibco, Carlsbad, CA) and 1% penicillin/streptomycin mixture were used to supplement culture medium. Cells were maintained under the standard condition that was set as the humidified atmosphere with 5% CO2 at 37°C. RPMI-1640 medium was replaced every 3 d. Cell passage was conducted after confluence reached 80%–90%. The culture methods were in accordance with the ATCC's guidance.
Cell transfection
To overexpress or silence the expression of SNHG5, U87 and U251 cells were transfected with pcDNA-SNHG5 or short hairpin RNA specific to SNHG5 (termed SNHG5 or sh-SNHG5). An empty pcDNA3.1 vector or nonspecific shRNA (termed NC or sh-NC) was used as a negative control. To enhance YY1 expression, cells were separately treated with pcDNA-YY1 and control (termed YY1 and NC). For suppressing YY1 expression, cells were separately treated with sh-YY1 and control (termed sh-YY1 and sh-NC). Cell transfections were performed in RPMI-1640 medium using Lipofectamine 2000 (Invitrogen) in line with user guide. All transfection plasmids were designed and synthesized by GenePharma Company (Shanghai, China). Forty-eight hours later, cells were harvested for further study.
RNA isolation and quantitative real-time polymerase chain reaction
TRIzol® reagent (Invitrogen) was utilized to isolate total RNA from cells based on the instructions of the supplier. The quantity and quality of total RNA were determined by NanoDrop 2000c (Thermo Scientific, Waltham, MA). RNA was subjected to reverse transcription reaction using Moloney Murine Leukemia Virus Reverse Transcriptase (M-MLVRT; Progema, Madison, WI). The complementary DNA (cDNA) was synthesized during the reaction. The RNA expression was measured using the One-Step SYBR® PrimeScript™ RT-PCR Kit (Perfect Real Time; TaKaRa Bio, Tokyo, Japan) and ABI 7500 system (Applied Biosystems, Carlsbad, CA). The specific primers were listed as follows: SNHG5, forward 5′- ACAATGGCGCTGTCTTCAGT-3′, reverse 5′-ACTCGTCCACACTCAGAACG-3′; YY1, forward 5′-GGAGACCATCGAGACCACAG-3′, reverse 5′- GCGCCCATCACACACATAAAA-3′; and GAPDH, forward 5′- GGGAGAAGCTGAGTCATGGG-3′, reverse 5′- TCCCGGTGACATTTACAGCC-3′. The PCR amplification protocol was described as follows: initiation incubation at 95°C for 5 min, followed by 40 cycles of melting at 95°C for 30 s, annealing at 60°C for 30 s, and final extension at 72°C for 1 min. The expression of RNA was calculated using the comparative 2−ΔΔCt method and ABI systems. GAPDH acted as an internal reference. PCR assay was performed in triplicate.
Cell counting kit-8 assay
The transfected U87 and U251 cells were seeded in a 96-well plate (1 × 103/well) with serum-free medium and cultured at 37°C with 5% CO2. Ten microliters of CCK-8 solution (CCK-8; Dojindo Molecular Technologies, Kumamoto, Japan) was added to each well and incubated for an hour at 37°C with 5% CO2. The OD value, relating to the number of active cells, was measured at 450 nm using microplate reader (Bio-Rad, Hercules, CA) at 1, 2, 3, and 4 d according to the standard method provided by the supplier. CCK-8 experiment was repeated at least three times.
EdU incorporation assay
EdU incorporation assay in U87 and U251 cells were conducted using the 5-ethynyl-20-deoxyuridine (EdU) Incorporation Assay Kit (Ribobio, Guangzhou, China) following the user guide. After transfection, cells were planted into 96-well plates and mixed with 100 μL of 50 μM EdU medium diluent for 3 h. After fixing in 4% paraformaldehyde, cells were cultured in 100 μL of the 0.5% Troxin X-100 for 10 min. Subsequently, cells were subjected to 100 μL of 1× Apollo® 488 fluorescent staining reaction liquid at 37°C for 30 min. Cell nucleus was stained by DAPI (Sigma, MO) in the dark. Experimental results were acquired from three independent replicates and showed as the ratio of the EdU-positive cells (red) to the total DAPI-positive cells (blue).
Flow cytometry of cell apoptosis assay
Flow cytometry was applied to detect GBM cell apoptosis rate in U87 and U251 cells. After different treatments, cells were collected and rinsed with precold phosphate-buffered saline (PBS; Invitrogen). Thereafter, cells were incubated with 100 μL of 1× Binding Buffer and double stained with the Annexin V Fluorescein Isothiocyanate (FITC)/Propidium Iodide (PI) Detection Kit (Thermo Fisher Scientific, Inc., Waltham, MA) for 15 min at 4°C. After incubation in the dark for half an hour, 400 μL of 1× Binding Buffer was added. Cells were analyzed by flow cytometry (BD Biosciences, San Jose, VA). Cell apoptosis rate was obtained in more than three independent experiments.
Caspase-3 activity detection
The caspase-3 activity was detected using the Caspase-3 Activity Kit (Beyotime Institute of Biotechnology, Guangzhou, China). First, cells were cultured in lysis buffer to obtain the total protein. After dilution, total protein was treated with caspase-3 substrate for 3 h. The free pNA (yellow formazan product) was released by caspase-3 to hydrolyze Ac-DEVD-pNA and tested at 405 nm. The caspase-3 activity was identified as the fold of enzyme activity compared with that of synchronous cells. Each procedure of experiment was repeated independently more than three times.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed in line with supplier's manual of the SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling Technology, CA). U87 and U251 cells were crosslinked in 4% formaldehyde for 30 min at room temperature. Glycine was added to a final concentration of 125 mM to halt the crosslinking reaction. Subsequently, cells were harvested and lysed in RIPA buffer. The lysates were cut by ultrasonic to generate DNA fragments of 200- to 1000-base pairs in length. The crosslinked DNA was immunoprecipitated with specific antibodies bound Dynabeads Protein G (Life Technologies, CA) overnight at 4°C. After washing, elution and decrosslinking, the recovered DNA was quantified using quantitative real-time polymerase chain reaction (qRT-PCR) with SYBR Green Mix (TaKaRa, Tokyo, Japan). IgG antibody was used as the negative control. Experimental procedures were performed at least three times.
Luciferase reporter assay
SNHG5 promoter luciferase assay was performed following the standard method of the Dual-Luciferase Reporter Assay System (Promega, Madison, WI). The wild-type and mutant form reporter plasmids of binding site 2 of YY1 within SNHG5 promoter region (termed site2-WT and site2-MUT) were constructed and purchased from GenePharma . U87 and U251 cells were cotransfected with site2-WT/MUT and pcDNA-YY1 together with an empty pcDNA3.1 vector (NC) in 24-well plates using Endofectin™-Plus (GeneCopoeia, Maryland). The reporter activity was measured by a Luciferase Assay Kit (Promega), normalizing to Renilla luciferase activity. Experimentation was performed more than three times.
Western blot
Cells were seeded into six-well plates to conduct western blot assay. The total protein was isolated from cells using RIPA buffer reagent (Thermo Fisher Scientific, Inc.). The concentration of the lysates was assessed using the Bicinchoninic Acid Kit (BCA; Thermo) based on the manufacturer's protocol. Following separation by sodium dodecyl sulfate/polyacrylamide gel electrophoresis, proteins were transferred onto polyvinylidene fluoride membranes and incubated on ice overnight. Thereafter, proteins were sealed in 5% bovine serum albumin for 2 h at room temperature. Membranes were incubated with primary antibodies (1:1000 dilution) specific to p38 (Abcam, Santa Cruz Biotechnology, Inc., Dallas, TX; ab31828), p-p38 (Abcam; ab4822), ELK1 (Abcam; ab32106), STAT1 (Abcam; ab30645), and GAPDH (Abcam; ab181602) at 4°C overnight and then rinsed gently three times with PBS (Invitrogen). Subsequently, the membranes were subjected to incubation with secondary antibodies (1:2000 dilution), which had conjugated to horseradish peroxidase for 2 h in the dark. Finally, all membranes were transferred to a freshly prepared enhanced chemiluminescence solution (ECL, Immobilon Western; Millipore, Billerica, MA). Protein bands were visualized. The protein levels of RNAs were normalized to GAPDH.
Cell treatment
U251 cell line was seeded into six-well plates and transfected with sh-SNHG5 or sh-NC for 48 h, followed by incubation with 10 ng/mL of TNF-α (Invitrogen) for 1 d. After transfecting with pcDNA-SNHG5 or an empty pcDNA3.1 vector, U87 cell was treated with 20 μM of SB203580 (Sigma Chemical Co., St. Louis, MO) for 24 h. Finally, U251 and U87 cell lines were reaped for subsequent analysis.
Bioinformatics analysis
The expression profile of SNHG5 in GBM tissue and normal tissues was acquired from The Cancer Genome Atlas (TCGA) database (
Statistical analysis
In this study, experimental results were acquired from at least three replicates. Statistical data were analyzed by SPSS® vision 22.0 software package (IBM, Chicago, IL) and expressed as mean ± standard error of mean (SEM). Two-tailed Student's t-test and one-way analysis of variance (ANOVA) were applied to compare the difference between groups. The threshold of statistical significance was set as probability lower than 0.05 (p < 0.05).
Results
SNHG5 was upregulated and promoted cell proliferation and inhibited apoptosis in GBM
With the help of TCGA dataset, they found that SNHG5 was highly expressed in GBM tissues compared with the normal tissues (Fig. 1A), indicating that the dysregulation of SNHG5 might be associated with the GBM tumorigenesis. Moreover, in comparison with the normal brain glial cell line HEB, they found the high expression level of SNHG5 in three human GBM cell lines (U251, U87, and LN229) (Fig. 1B), among which U251 exhibited the highest expression of SNHG5 and U87 the lowest. With that, SNHG5 was overexpressed by pcDNA-SNHG5 (termed SNHG5) in U87 cell line and silenced by the shRNA specific to SNHG5 (termed sh-SNHG5) in U251 cell line (Fig. 1C). Results of CCK-8 assay displayed that an increased viability of U87 cells with the presence of SNHG5 overexpression, compared with NC group transfected with the empty vector (Fig. 1D left). The viability of U251 cells in sh-SNHG5 group was obviously lower than that in sh-NC group (Fig. 1D right). Results of EdU assay demonstrated that proliferative ability of GBM cells was enhanced by SNHG5 overexpression, but reduced in response to the interference of sh-SNHG5 (Fig. 1E). Besides, they applied flow cytometry analysis to measure GBM cell apoptosis under the overexpression and knockdown of SNHG5. As illustrated in Figure 1F, the apoptotic cells were obviously lessened by SNHG5 overexpression, but were markedly increased by SNHG5 knockdown. Caspase-3 activity was a positive indicator of cell apoptosis. 30 In this study, they found that the silenced SNHG5 expression resulted in an augmented activity of caspase-3, in contrast, the strengthened SNHG5 expression repressed caspase-3 activity (Fig. 1G). Altogether, the data above indicated that SNHG5 was upregulated and promoted cell proliferation and inhibited apoptosis in GBM.
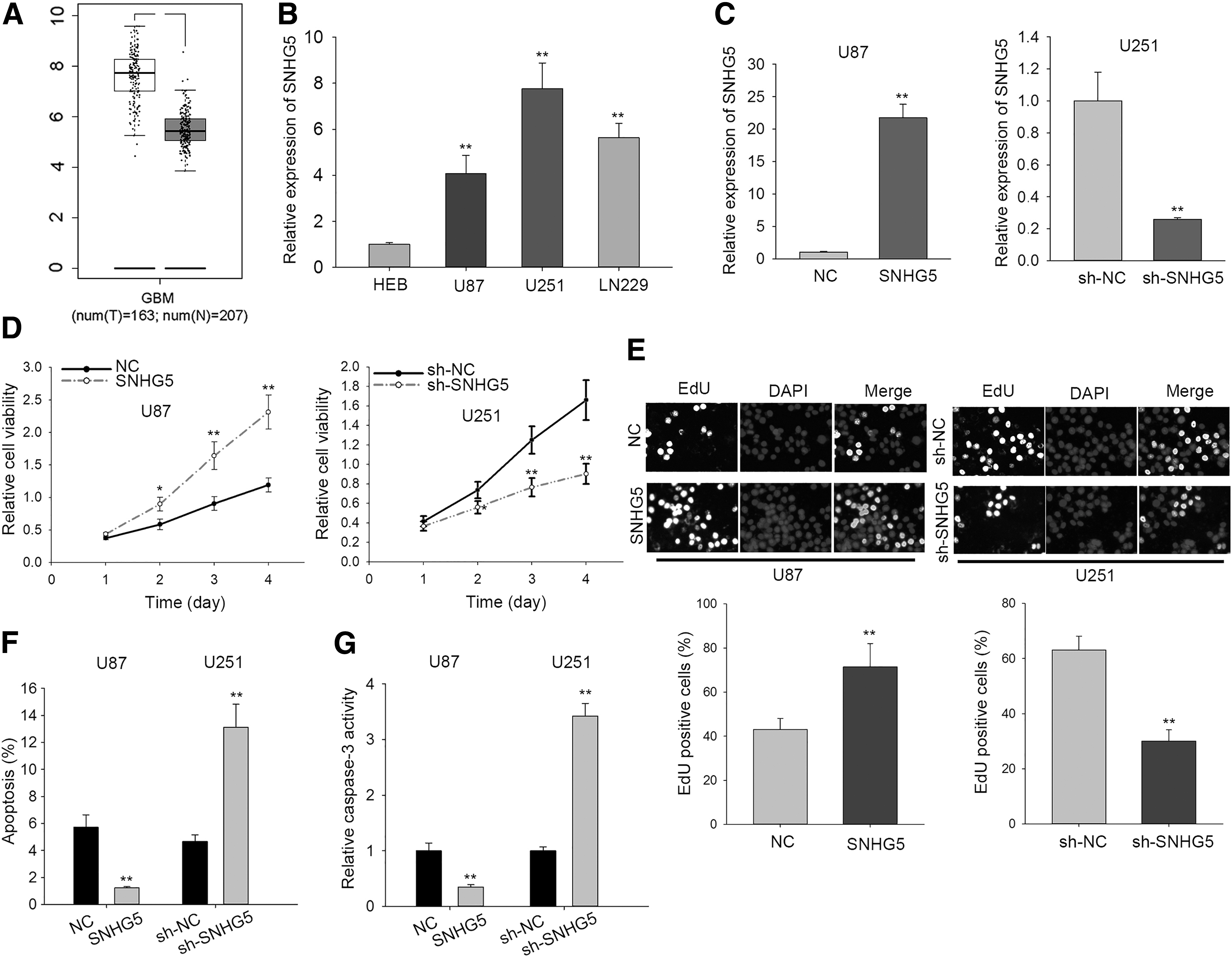
The upregulation of SNHG5 promoted GBM cell proliferation.
YY1 acted as a transcriptional activator of SNHG5
These above results prompted the authors to explore the molecular mechanism which contributed to the upregulation of SNHG5 in GBM. Recently, transcription factors were reported to activate their target gene expression in various tumors. 31 –33 UCSC tool revealed that Yin Yang 1 (YY1) was one of the potential transcription factors of SNHG5. It is well known that YY1 can induce target gene expression in various cancers at the transcriptional level. 34,35 For this reason, the authors chose YY1 for subsequent analysis. Expectedly, they identified several putative binding sites of YY1 within SNHG5 promoter region using JASPAR prediction tool. The motif and fragments containing top two putative binding sites (indicated as P1 and P2) were obtained from JASPAR (Fig. 2A). ChIP assay revealed that only the P2 fragment containing binding site 2 was responsible for the binding of YY1 to SNHG5 promoter region (Fig. 2B). Afterward, they overexpressed YY1 expression in both U87 and U251 cell lines (Fig. 2C). Luciferase reporter assay was utilized to verify the binding of YY1 to binding site 2. Experimental result showed that the luciferase activity of binding site 2 wild type (indicated as site2-WT) was enhanced upon YY1 overexpression in U87 and U251 cell lines, but that of binding site 2 mutant form (indicated as site2-MUT) was not altered by YY1 overexpression (Fig. 2D). RT-qPCR and western blot assays indicated that the mRNA and protein expression of YY1 was increased in GBM cell lines in comparison to normal cell line HEB (Fig. 2E), which was consistent with SNHG5. Additionally, the enhanced YY1 expression activated SNHG5 transcription in both U87 and U251 cell lines and knockdown of YY1 had opposite effects (Fig. 2F). Therefore, these results indicated that YY1 could bind with SNHG5 promoter to accelerate its transcription.
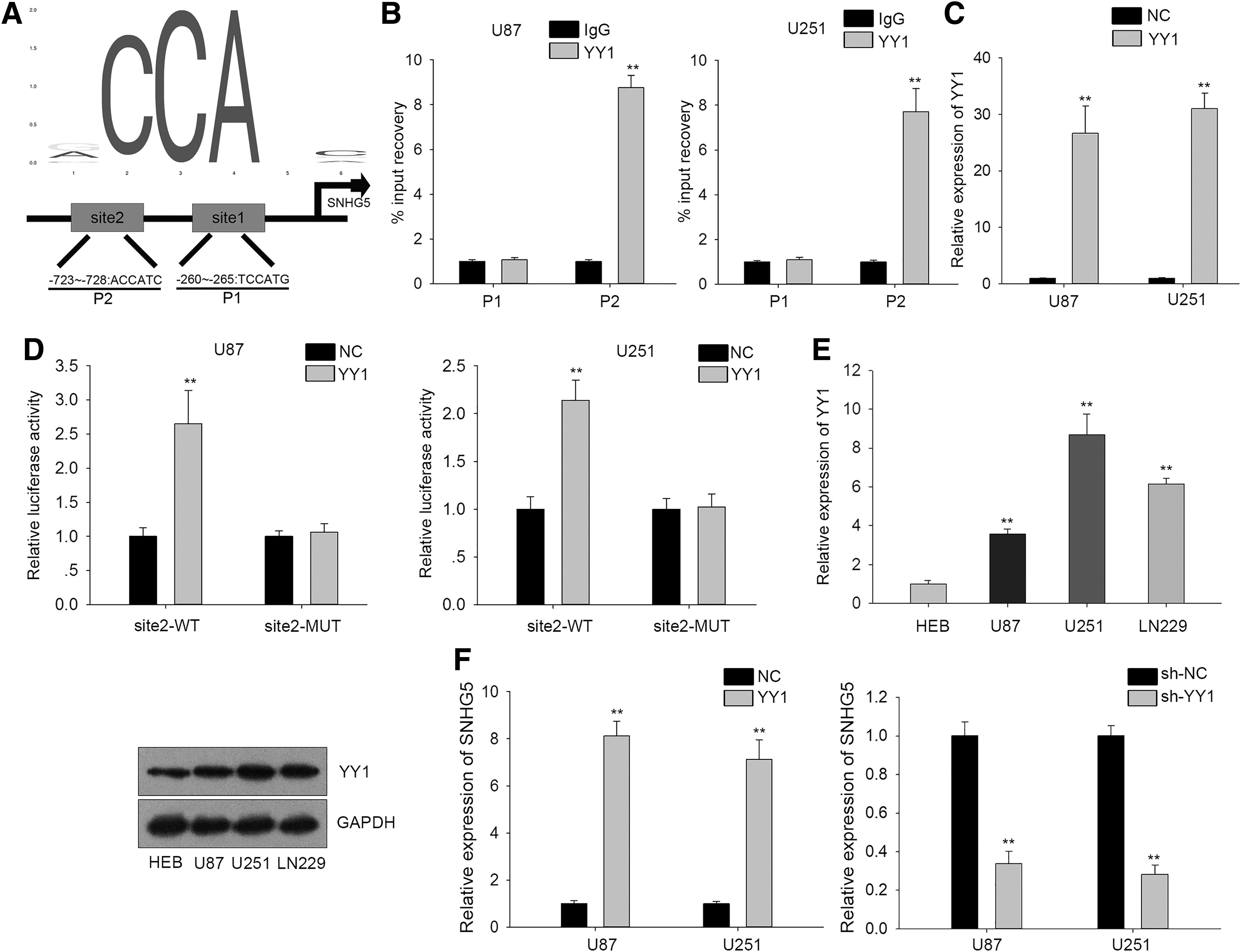
YY1 acted as a transcriptional activator of SNHG5.
Activating p38/MAPK signaling pathway recovered the GBM cell proliferation inhibited by SNHG5 knockdown
Based on the previous study, 29 p38/MAPK signaling pathway could involve in the lncRNA-mediated GBM development. To find out whether SNHG5 regulated GBM progression through p38/MAPK pathway, the authors first evaluated the impact of SNHG5 on p38/MAPK signaling pathway by detecting the protein levels of phosphorylated p38 (termed p-p38) and total p38 (termed p38) in response to SNHG5 overexpression or knockdown. Results of western blot indicated that the protein level of p-p38 was strengthened by SNHG5 overexpression and weakened by SNHG5 knockdown, but there was no obvious change in protein level of the p38 (Fig. 3A). Importantly, the addition of p38/MAPK pathway activator TNF-α (10 ng/mL) counteracted the inhibitory effect of SNHG5 knockdown on the protein level of p-p38 and pathway downstream genes (ELK1 and STAT1) (Fig. 3B). As displayed in Figure 3C, D, the proliferative capacity of U251 cells was notably inhibited by silencing SNHG5 expression, but was reversed by TNF-α. The significant increase in caspase-3 activity evoked by the intervention of sh-SNHG5 was recovered in part by TNF-α in U251 cell line (Fig. 3E).

TNF-α recovered the GBM cell proliferation inhibited by SNHG5 knockdown.
Suppressing p38/MAPK signaling pathway abrogated the GBM cell proliferation promoted by SNHG5 overexpression
Additionally, they also inhibited p38/MAPK pathway through SB203580 (20 μM) to examine whether SNHG5 regulated GBM progression through p38/MAPK pathway. Results from western blot assay suggested that the increased protein levels of p-p38, ELK1, and STAT1 caused by SNHG5 overexpression were repressed by SB203580 (Fig. 4A). CCK-8 and EdU assays exhibited that SB203580 impaired the promotive effect of SNHG5 overexpression on U87 cell proliferation (Fig. 4B, C). By detecting caspase-3 activity, they observed that the obvious decrease in caspase-3 activity in U87 cell was caused by the transfection with pcDNA-SNHG5, whereas, the blockade of p38/MAPK pathway dramatically reversed this descending activity in U87 cell apoptosis (Fig. 4D). Taken together, these results suggested that SNHG5 regulated GBM cell proliferation through p38/MAPK signaling pathway.
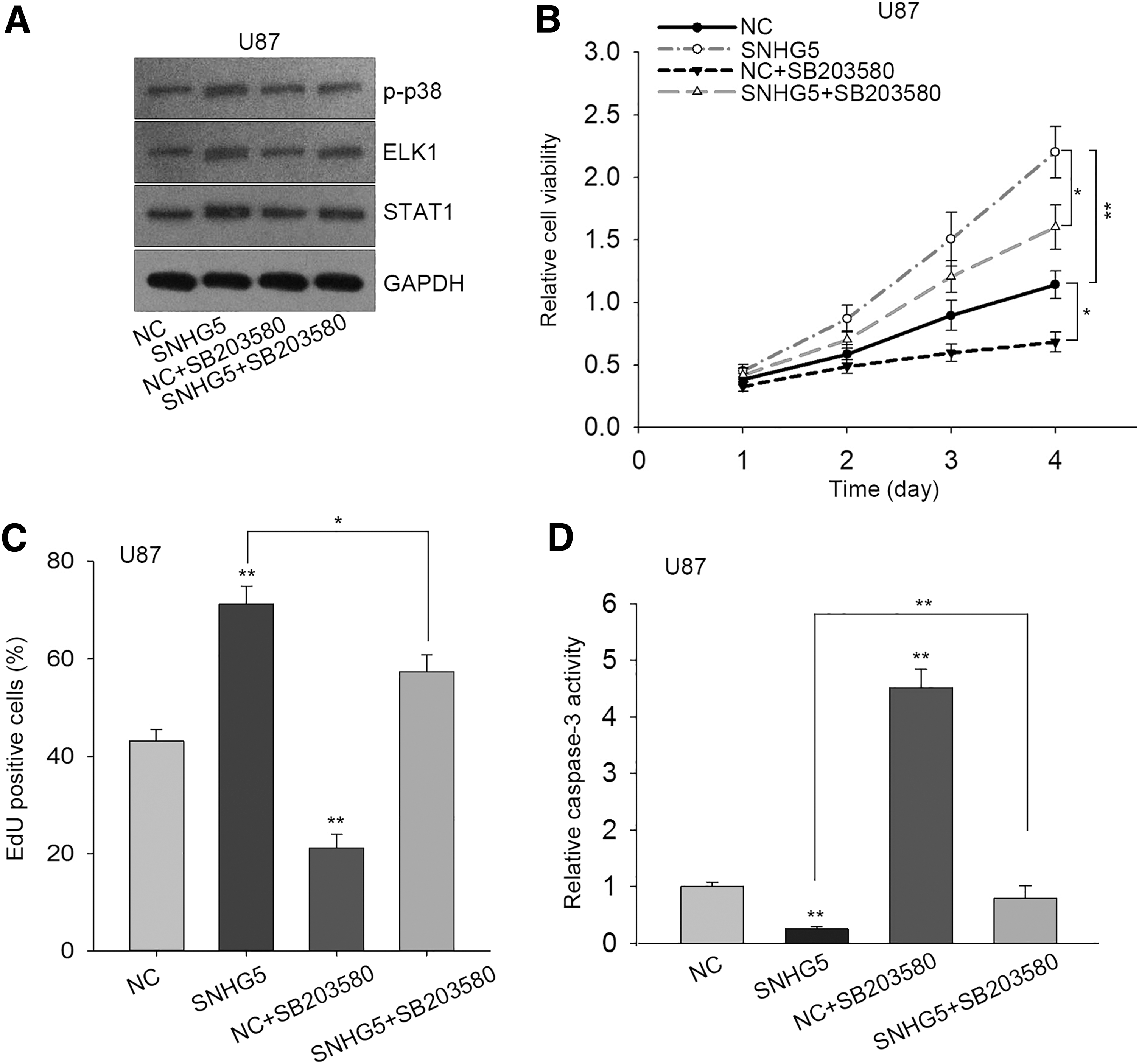
SB203580 abrogated the GBM cell proliferation promoted by SNHG5 overexpression.
Discussion
GBM is one of the most common and aggressive types of human primary brain tumors. Statistics show that GBM accounts for 52% of solid brain tumor and 20% of intracranial tumors. 36 Despite that the mechanisms contributing to occurrence and progression of GBM were extensively investigated in the past years, the pathogenic mechanism of GBM remains ill defined. Moreover, the regulatory role of genes in GBM is still unknown in a wide range.
An increasing number of evidences indicate that lncRNAs exert a crucial role in regulating tumorigenesis of GBM. For instance, lncRNA TUG1 was identified to remarkably promote cell proliferation and tube formation as well as reduce angiogenesis ability in GBM 37 ; knockdown of SOX2OT repressed GBM cell proliferation and accelerated cell apoptosis. 38 It is evident that cell proliferation is an important stage in initiation and development of GBM. Therefore, they principally aimed to investigate the effect of lncRNA's dysregulation on GBM's cell proliferative capacity. In the present study, the authors first determined the upregulated expression of SNHG5 in GBM. Afterward, they carried out CCK-8 and colony formation assays to evaluate the effect of SNHG5 on GBM cell proliferation, as well as flow cytometry and caspase-3 activity detection to assess cell apoptosis. Results of functional experiments revealed that SNHG5 promoted cell proliferation and suppressed cell apoptosis in GBM.
The upregulation of SNHG5 prompted them to investigate the mechanism responsible for the activation of SNHG5 in GBM. Influenced by the notion that transcription factor YY1 induces lncRNA expression in various cancers, 39,40 they applied UCSC to determine whether YY1 was the putative transcription factor of SNHG5. JASPAR tool was utilized to acquire the potential binding sites of YY1 in the SNHG5 promoter region. Mechanism assays validated that YY1 functioned as a transcriptional activator of SNHG5. In general, the expression profile of transcriptional activator was positive with its target gene in the same tumor. In expectation, qRT-PCR disclosed the upregulation of YY1 in GBM.
Generally, signaling transduction pathways are involved in lncRNA-mediated cancerous development, including p38/MAPK signaling pathway. 41,42 Western blotting results demonstrated that SNHG5 promoted p38 protein level under phosphorylation, indicating that SNHG5 could activate p38/MAPK pathway in GBM. Finally, rescue assays were designed with the treatment of p38/MAPK pathway activator TNF-α or inhibitor SB203580. Results suggested that SNHG5 regulated GBM development through p38/MAPK pathway.
As far as they know, their study is the first to identify the oncogenic role of SNHG5 in GBM. SNHG5 promoted GBM cell proliferation and inhibited apoptosis. Mechanistically, YY1-induced SNHG5 accelerated GBM cell proliferation through activating p38/MAPK pathway. However, the detailed mechanism whereby SNHG5 regulates p38/MAPK pathway needs more exploration. Despite the authors' findings are preliminary, they are promising to set the future study direction.
Footnotes
Acknowledgment
Authors appreciate each member who contributed to this study.
Disclosure Statement
Authors declare that there are no conflicts and interests.
Funding Information
This study was supported by grants from Hunan Provincial Health and Wellness Committee Research Project (NO. C2019053, NO. C2019054).