
Abstract
Objectives:
The epidermal growth factor receptor (EGFR) signaling pathway is widely recognized to play an important role in development and progression of many malignant tumors, including lung cancer. The aim of this study was to produce a useful technetium-99m (99mTc) complex for early detection and staging of EGFR-positive tumors.
Methods:
The authors labeled quinazoline derivative (3-iodinephenyl) quinazoline-4, 6-diamine with 99mTc using S-acetylmercaptoacetyltriglyglycinate (MAG3) conjugated to quinazoline derivative through 4-[(2-aminoethyl)amino]-4-oxobut-2-enoic acid because it is a bifunctional chelator with an average yield of 93.9% ± 2.5% and radiochemical purity of >98%.
Results:
In vitro studies indicate that the [99mTc]-complex13 has high stability in physiological conditions and binds the HCC 827 cells and shows HCC 827 internalization. The biodistribution of the [99mTc]-complex13 in healthy Institute of Cancer Research mice indicates the rather fast blood clearance through the hepatobiliary system.
Conclusion:
These preliminary results pave the road for estimating the status of EGFR and monitoring the response to EGFR tyrosine kinase inhibitors as a personalized cancer therapy regimen.
Introduction
Epidermal growth factor receptor (EGFR) is part of the human EGFR family. It contains an extracellular ligand binding domain, a transmembrane region, and an intracellular protein cytoplasmatic domain with tyrosine kinase activity. 1,2 Ligand binding to EGFR triggers dimerization of two EGFRs, followed by phosphorylation of tyrosine residues in intracellular cytoplasmatic domain and then activation of downstream signaling that leads to cell proliferation and protection from apoptosis. 3 Previous reports showed overexpression of EGFR in various cancers such as head and neck tumors, colorectal, breast, non-small-cell lung cancer, and many types of epithelial cancers. 4 Furthermore, the crucial importance of EGFR to tumor progression and resistance to other treatments has motivated research on novel treatment strategies to target these receptors. 5,6
Monoclonal antibodies directed against the extracellular ligand binding domain of the receptor and small molecule tyrosine kinase inhibitors (TKIs) directed against the intracellular adenosine triphosphate (ATP) binding site of the tyrosine kinase domain are two classes of drugs approved for clinical use for targeting EGFR. 7 –10 However, the radiolabeled antibodies clear slowly from circulation which causes dose-limiting hematologic toxicity and requires a longer wait time between injection of the radiolabeled antibody and imaging, 11 whereas the TKIs clear quickly from blood, offering a more flexible time window for imaging.
The superiority of the two commonly used EGFR TKIs (i.e., gefitinib and erlotinib) over conventional chemotherapy has been reported, and there has also been growing interest in using radionuclide (e.g., 18 F, 11 C, 99mTc, and 111In)-labeled TKIs as biomarkers 12 –14 for targeting EGFR. 15 –18 Several classes of TKIs have been established, among which those based on the 4-anilinoquinazoline core scaffold analogues were identified as the most potent competitive inhibitors of ATP, and they are the most heavily investigated so far. 19,20 Several EGFR TKIs have been approved as curative effective inhibitors for targeting EGFR by the U.S. Food and Drug Administration (FDA); however, the clinical effects of these inhibitors vary in many cancers because of the different levels of expression and mutations of EGFR because of the heterogeneity of cancers. 21
Although radiotracers with the potential ability to reflect the status of EGFR have been developed, some inadequacy was observed. For example, 18 F-PEG6-IPQA has a complicated synthesis procedure and the 18 F-PEG6 metabolic cleavage from the quinazoline core will increase the background signal. 22 11 C-gefitinib has substantial uptake in the intestine, and the application is hampered by nuclide 11 C, 23 for its short half-life. In addition, it is not convenient for PET studies because of the need for an on-site cyclotron.
The widespread availability of single-photon emission computed tomography (SPECT) scanners and 99Mo-99mTc generators has suggested that the development of metastable technetium-99m (99mTc)-labeled molecular probes offers a distinct advantage for imaging. The traditional method of 99mTc-labeling biomolecules involves using N-hydroxysuccinimidyl hydrazino nicotinate hydrochloride (NHS-HYNIC) 24 and requires a coligand. The resulting chelate usually has poor in vivo and in vitro stability. 25 Here, the authors used the N-hydroxysuccinimidyl S-acetylmercaptoacetyltriglyglycinate (NHS-MAG3) as a bifunctional chelator that is easily synthesized and conjugated. The conjugation product was easily radiolabeled with 99mTc, and the radiolabeled probes have shown stability suitable for diagnosis and therapeutic application in the laboratory. 26
According to the previous researches, 19,20,27 4-anilinoquinozoline quantitative structure–activity relationship could be summarized as follows: First, the nitrogen atom at 1-position forms a hydrogen bond with the key amino acid Met-769 or Met-793, which is important for the combination of 4-anilinoquinozoline to the EGFR target. For the same reason, the N-3 atom by acting as a hydrogen-bond acceptor to interact with the target is also important for its inhibitory potency. Second, the hydrophobic substituent group at 4-position stretch into a hydrophobic cavity that contains a variety of amino acids and opens at Thr790. The interaction of the hydrophobic substituent group with the hydrophobic cavity confers high avidity to the inhibitor and the target. In addition, the halogen substituted anilines at the C-4 position are essential for improving its inhibitory potency. Finally, the Michael acceptor side chain of irreversible inhibitors at 6-position forms a covalent bond with the sulfhydryl group of the Cys-773 of EGFR. Molecular modeling studies have indicated that a covalent bond confer a greater affinity than the reversible inhibitor.
Based on this information, the authors reported the synthesis and characterization of a MAG3-conjugated quinazoline analogue and the synthesis, characterization, and biological evaluation of the corresponding 99mTc complex13.
Materials and Reagents
99mTc-pertechnetate was purchased from Shanghai Atom Kexing Pharmaceutical Co., Ltd. (Shanghai, China) and eluted in saline solution. SnCl2·2H2O were purchased from Sigma-Aldrich Corporation (St. Louis, MO). All other solvents and reagents were analytical grade from China National Pharmaceutical Group Corporation (Beijing, China) and used without further purification. The C18 Sep-Pak columns were purchased from Huayi Isotopes (Jiangsu, China).
Human lung cancer cell line (HCC 827) was obtained from the Cell Bank of Type Culture Collection of Chinese Academy of Sciences (Shanghai, China). Institute of Cancer Research (ICR) male mice (6–8 weeks old) were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China) which were housed under specific pathogen-free conditions at the Department of Laboratory Animal Science of Fudan University (Shanghai, China). All procedures were performed in accordance with guidelines approved by the Animal Care Committee of Fudan University (Shanghai, China).
Synthesis of 12
The synthesis of
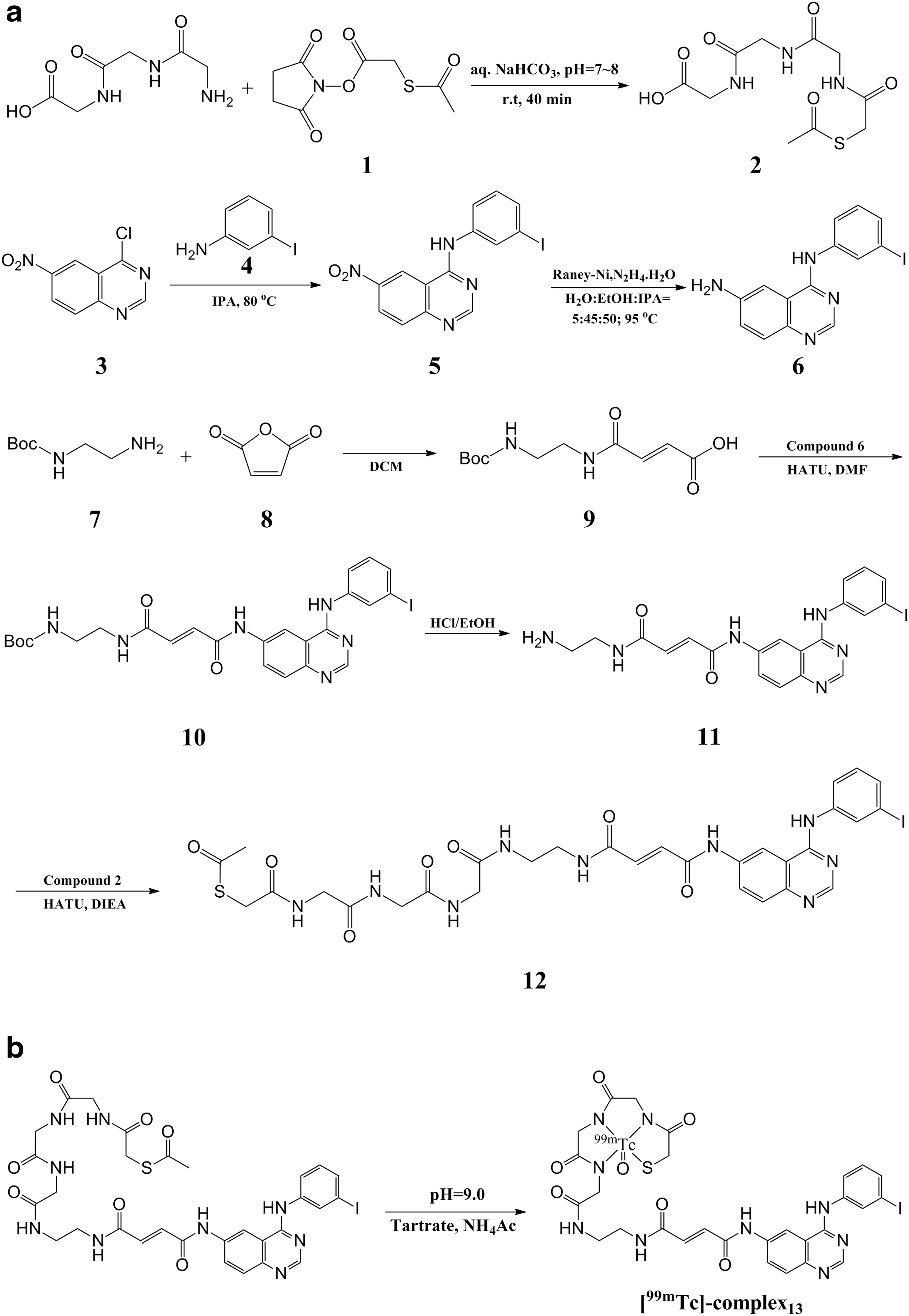
A solution of compound 7 (2.00 g, 12.48 mmol, 1.96 mL),
A solution of
In vitro radiolabeling
The synthesis of [99mTc]-complex13 is shown in Figure 1b. In brief, 20 μg of compound 12 was dissolved in 100 μL DMF, and then the mixture was added to a combined solution of 45 μL ammonium acetate (0.25 M) and 15 μL tartrate buffer, and subsequently <100 μL (∼370 MBq) freshly generated
In vitro stability study
After purification, [99mTc]-complex13 was diluted and incubated in 0.01 M phosphate-buffered saline (PBS), normal saline, and 0.1% BSA for 1, 2, 3, 6, and 9 h at 37°C. After incubation, the samples were filtered using a 0.22 μm Millipore filter and assessed by RP-HPLC (Agilent technologies 1260 Infinity with a variable wavelength UV detector and a gamma detector; HPLC column: phenomenex Gemini, 5 μm NX-C18 110 Å, 150 × 4.6 mm; mobile phase: 0.1% TFA in H2O, 0.1% TFA in acetonitrile eluting with a gradient flow over 30 min of 1:99 H2O/MeCN to 20:80 H2O/MeCN, flow rate: 1 mL/min).
Cellular uptake studies
In brief, HCC 827 cells were cultured with RPMI 1640 cell culture medium supplemented with 10% fetal bovine serum and antibiotic mixture (100 U/mL penicillin and 0.1 mg/mL streptomycin), and incubated at 37°C in humidified air with 5% CO2. Cells were kept in the log-phase of proliferative activity. For cell binding studies, HCC 827 cells (5 × 105 cells/mL per well) were seeded in 24-well plate and incubated for 48 h. To prevent EGFR activation, HCC 827 cells were washed with culture medium without fetal bovine serum three times. Then, the cells were incubated with [99mTc]-complex13 (0.925 MBq/well) with or without the blocking precursor (1000-fold molar of compound 12) for different time intervals (5, 30, 60, 120, and 240 min). The total volume of each well was 0.5 mL. Cells were washed three times with PBS and the supernatants were collected, then the cells were detached with 1.0 M NaOH. The radioactivity of supernatant and cells was measured by γ-counter (CRC-15R; Capintec, Inc., Ramsey, NJ). The cell uptake of [99mTc]-complex13 was expressed as a percentage of the total added radioactivity with decay corrected. All experiments were conducted in triplicate to ensure reproducibility.
Biodistribution studies
Eighteen healthy male ICR mice (6–8 weeks old; weighing 18–20 g) were randomly selected for in vivo biodistribution study. Then, the mice were injected through the tail vein with 150 μL of 5 MBq [99mTc]-complex13. The mice were randomly grouped and killed at 5, 15, 30, 60, and 180 min with slightly overdosing anesthesia (0.1 M chloral hydrate, 0.2 mL/mouse) postinjection. Blood, heart, liver, spleen, lung, kidney, brain, muscle, stomach, bone, skin, small intestine, large intestine, and thyroid were harvested and placed into weighed tubes. The radioactivity was measured using an automatic gamma counter and uptake was calculated as the percentage of the total injected dose of radioactivity per gram of tissue (%ID/g), and data were plotted as histograms.
Results
Chemistry
As shown in Figure 1a, 2 was prepared from
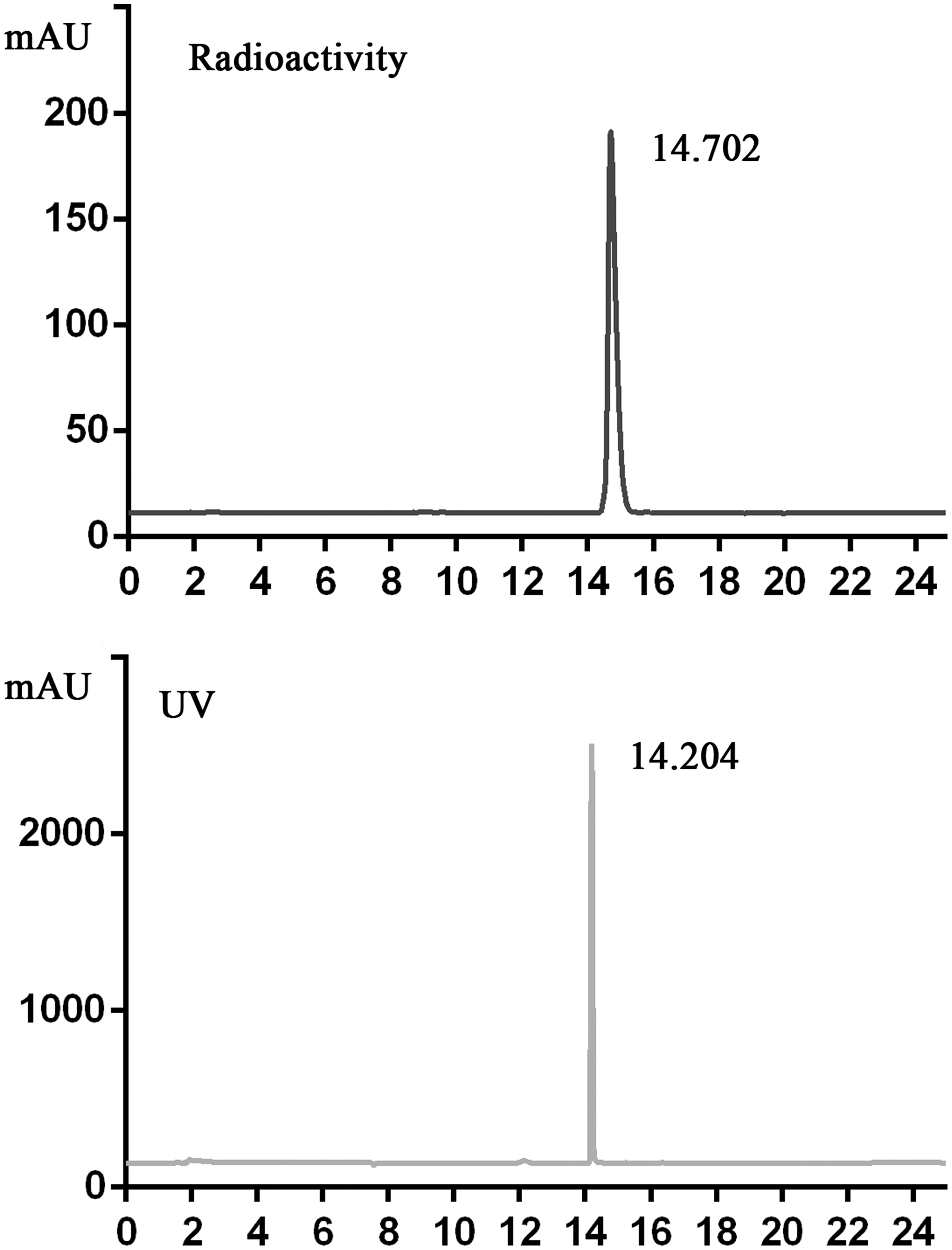
Analytical HPLC chromatogram of [99mTc]-complex13 co-injected with complex13. Black: Radioactive detection. Gray: UV detection. There was a small difference in retention time because of the setup of a serial detector. HPLC, high-performance liquid chromatography.
Radiochemistry
The synthesis of [99mTc]-complex13 is shown in Figure 1b. A single-step method followed the developed method with a few modifications.
26
In brief, the precursor was dissolved in labeling buffer and reacted with
In vitro stability
Samples of [99mTc]-complex13 were examined for its in vitro stability, and the analyses were repeated three times. As shown in the radio-HPLC results at different points in time, [99mTc]-complex13 demonstrated pronounced stability, and there was no decomposition observed in PBS, normal saline, or BSA incubation after incubation at different points in time from 1 to 9 h, suggesting excellent stability in vitro (Fig. 3).

Stability analysis of [99mTc]-complex13 in 0.01 M PBS (left panel), 0.9% NaCl (middle panel), and 0.1% BSA (right panel) at 1, 2, 3, 6, and 9 h, respectively. PBS, phosphate-buffered saline.
Cellular uptake studies
The cellular uptake of [99mTc]-complex13 was evaluated using HCC 827 cells with a modified version of a previously described method, 28 and the results were plotted. As given in Figure 4, [99mTc]-complex13 showed a rapid uptake by the HCC 827 cells during the initial phase (the first 60 min). At the time point of 60 min, the accumulation of [99mTc]-complex13 in HCC 827 cells had reached at 1.36% ± 0.11% (n = 3). Addition of the precursor (1000-fold molar of compound 12) significantly decreased the internalization of the [99mTc]-complex13 in HCC 827 cells. After the first 30 min, the internalization of [99mTc]-complex13 leveled off at 0.282% ± 0.019% (n = 3).
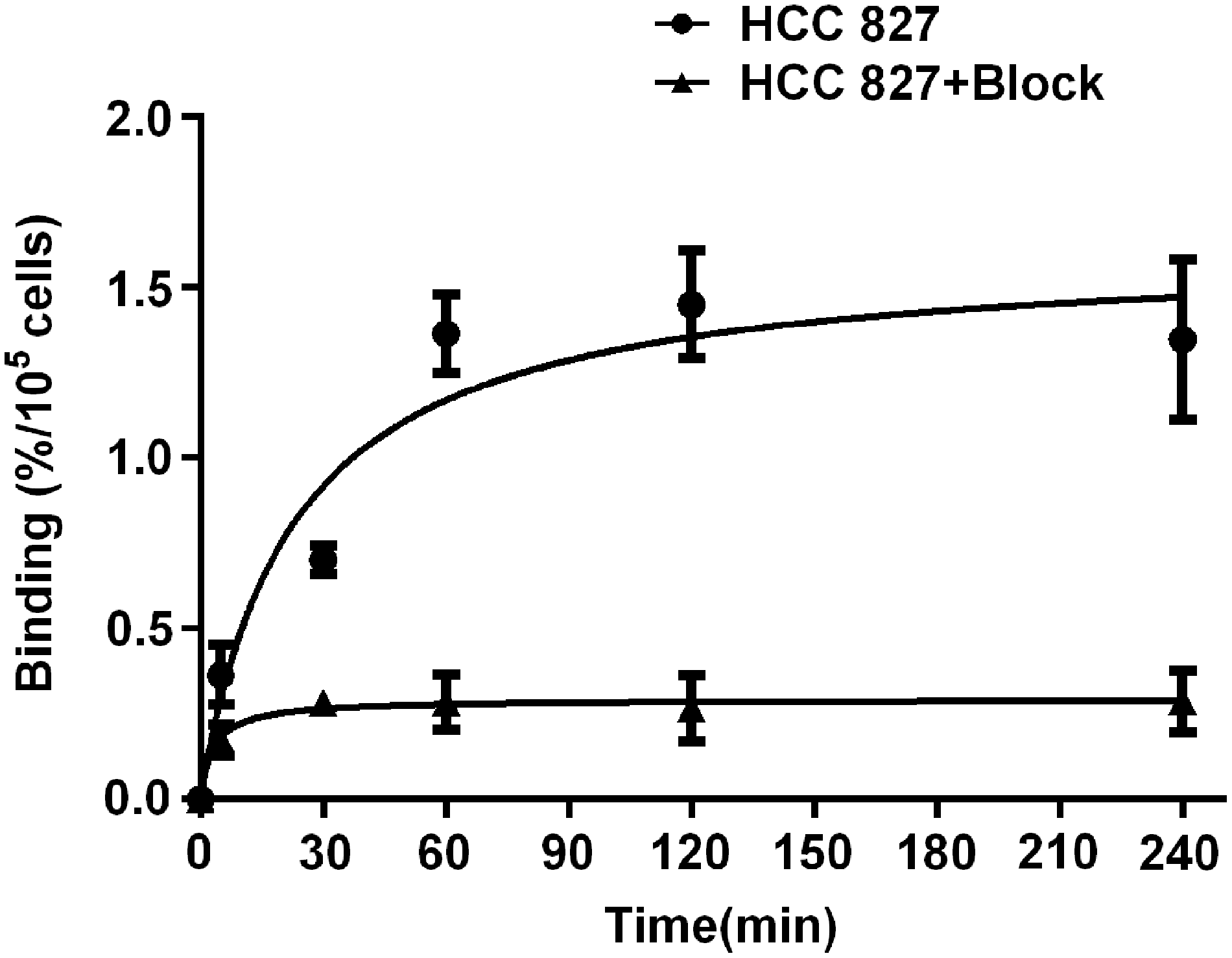
Uptake of [99mTc]-complex13 into HCC 827 cells were measured at the indicated points in time.
Biodistribution studies
To examine the in vivo stability of [99mTc]-complex13 and its pharmacokinetic profile, the biodistribution in healthy female ICR mice was tested after injection at different points in time (Fig. 5). The blood concentration decreased from 11.36 ± 0.67%ID/g at 5 min post injection to 0.26 ± 0.15%ID/g at 15 min postinjection, which showed rather rapid blood clearance. High probe uptake in the liver was observed at 15 min postinjection with a value of 20.92 ± 1.65%ID/g, but the probe cleared quickly to 0.99 ± 0.52%ID/g at 30 min postinjection. The accumulation of the probe within the wall of the small intestine increased to 28.04 ± 1.34%ID/g at 15 min postinjection and then decreased slowly to 3.72 ± 1.03%ID/g at 60 min postinjection. Accumulation in the kidney lasted for 60 min postinjection. The activity in the two lungs was 25.49 ± 6.28%ID/g at 5 min postinjection, which was followed by a gradual clearance over time (from 15 to 60 min postinjection). The activity in other organs such as the heart, spleen, brain, muscle, stomach, skin, thyroid, and large intestine remained low throughout the study. The uptake of the probe by all the organs decreased to no more than 0.072 ± 0.015%ID/g (except for the great intestine: 0.689 ± 0.076%ID/g) at 180 min postinjection.

Biodistribution (mean ± SD, n = 3) of radioactivity (%ID/g) in the ICR mice after injection of [99mTc]-complex13. ICR, Institute of Cancer Research; SD, standard deviation.
Discussion
Over the past decades, the EGFR has emerged as an effective target for tumor therapy. Multiple molecular imaging probes have been developed to assess EGFR expression level in tumors, mainly including antibody-based and peptide-based imaging agents. However, because of the inherent limitations of these agents such as relatively large size, weak tissue penetration, and unfavorable receptor binding kinetics, imaging applications with these molecules have been limited. 6,19 The tailored drugs have smaller size, which offer many advantages in molecule imaging, for example, more favorable pharmacokinetics, better tissue penetration, and more preferable flexibility in chemical modification. 25,29
To date, several EGFR-targeted probes have been reported in evaluating the EGFR expression, such as the carbon-11, fluorine-18, or iodine-125-labeled anilinoquinazoline derivatives, which have shown promise in imaging EGFR expression level in nonsmall cell lung carcinoma (NSCLC) patients. 9,22,30 Whereas the use of 11 C and 18 F radionuclides in clinical trials have been hampered to a certain extent because of their short half-life, which require an on-site cyclotron for production, and repeated fabrication (especially 11 C) when several subjects need to be examined, the 125I has a long half-life that would promote accumulation radiation dosimetry. Considering widespread application, in this study, an anilinoquinazoline derivative was conjugated with a bifunctional chelator and then labeled with 99mTc to establish a novel SPECT imaging probe, namely [99mTc]-complex13. 99mTc is a commonly used radionuclide in the clinic, with appropriate half-life and excellent radiochemical properties for nuclear imaging.
The radiolabeling of compound12 was rapid and efficient, with a high RCY rate, which was consistent with the convenient formulation of the labeling kit. In addition, the in vitro stability of [99mTc]-complex13 was favorable in PBS and saline that could last at least for 9 h. Although [99mTc]-complex13 was quickly bound by the albumin when incubated with 0.1% BSA, this physical adsorption did not affect the stability of [99mTc]-complex13.
HCC 827 cell line has been characterized as a sensitive one to gefitinib, which is another EGFR inhibitor sharing an identical 4-anilinoquinozoline core scaffold with compound 12. To evaluate the potential utility of [99mTc]-complex13 as a molecular probe for EGFR-TK imaging, the authors carried out in vitro cellular studies to obtain first insight into the internalization of the [99mTc]-complex13 in HCC 827 cells at different points in time. As given in Figure 4, the results indicated that the probe bound specifically to the EGFR-expressing cells.
The biodistribution of [99mTc]-complex13 was measured in healthy ICR mice at 5, 15, 30, 60, and 180 min postinjection. As given in Figure 5, the most striking feature of the [99mTc]-complex13 was the fast clearance from the bloodstream and excretion from the major organs such as the liver, lung, and intestines, which substantially reduced background radioactivity. Of note, because of physical adsorption, a few radioactive particles with diameters larger than those of the capillary might generate. When the [99mTc]-complex13 solution was injected into the vein, embolism might occur in the pulmonary microvasculature. But it would be substantially different from the 99mTC-MAA used clinically, the activity of which remained detectable for 2 d after injection, whereas the [99mTc]-complex13 could be cleaned fast from the pulmonary with no significant activity 1 h later, similar to the result of the previous study. 18 This might be because the aggregation was caused by simple physical adsorption rather than the chemical bond formation.
The main excretory route of the [99mTc]-complex13 was the hepatobiliary system, as evidenced by decreasing activity in liver and accumulation of radioactivity in the mouse feces (88.4 ± 1.5%ID/g at 24 h postinjection, data not shown). The activities of the stomach and thyroid were consistently low, which indicated no in vivo reoxidation to
Clinical studies have shown that the high expression of EGFR kinase was associated with improved progression-free survival and overall survival after treatment with EGFR kinase inhibitors compared with patients with wild-type EGFR. Furthermore, it is unfeasible to administrate repetitive invasive punctures of multiple tumor lesions in patients with advanced NSCLC. 11,22 However, the whole-body noninvasive imaging based on probes specific for activated status of EGFR kinase could provide information of EGFR expression of the tumor lesions for forecasting treatment response before initiating the EGFR inhibitor-based therapy. 34 The results demonstrated the feasibility of using complex13 as a SPECT tracer for screening EGFR TKIs sensitive patients. In addition, complex13 holds promise not only for noninvasively screening sensitive patients for treatment of EGFR inhibitors but also for guiding tumor excision. This would bring remarkable benefits to patient's therapy response and improve the survival rate.
Conclusion
In this study the authors evaluated a highly efficient chemical synthesis of [99mTc]-complex13, which produced the 99mTc labeling of the quinazoline scaffold in high yield and with high radiochemical purity. Animal studies indicated a high in vivo stability and a fast clearance from the circulation. Biodistribution studies of [99mTc]-complex13 in healthy mice have been favorable. Taken together, preliminary biological studies have been carried out, and in vivo studies in tumor-bearing mice are warranted to prove its feasibility as an imaging probe for early detection and staging of EGFR-positive tumors. Altogether, the results suggest that the tracer has the potential to be used as a SPECT probe to identify EGFR TKI sensitive patients.
Footnotes
Authors' Contributions
Z.S., Methodology; P.H., Methodology; J.Z., Software; Q.L., Software; Y.X., Design; D.C., Design. All authors have reviewed the article and approved publication before submission.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (No. 81671735 and 81471706), the Open Large Infrastructure Research of Chinese Academy of Sciences, the Shanghai Science and Technology Committee International Collaboration project (16410722700) and Shanghai Sailing Program (17YF1417400).