
Abstract
Background:
Hepatocellular carcinoma (HCC) is associated with high morbidity and mortality and has become the most frequently diagnosed liver cancer globally. Long noncoding RNAs have been widely studied because they exert essential functions in human diseases.
Aim of the Study:
The aim of the study is to explore the role and molecular regulatory mechanism of TRIM52-AS1 in HCC.
Materials and Methods:
Real-time quantitative polymerase chain reaction examined TRIM52-AS1, miR-514a-5p, and mitochondrial ribosomal protein S18a (MRPS18A) expression in HCC cells. Cell Counting Kit-8 (CCK-8), colony formation, flow cytometry, JC-1, transwell, and Western blot assays uncovered the function of TRIM52-AS1 in HCC. RNA immunoprecipitation (RIP), RNA pull down, and luciferase reporter assays validated the association among TRIM52-AS1, miR-514a-5p, and MRPS18A. Nuclear–cytoplasmic fractionation assay revealed the subcellular location of TRIM52-AS1 in HCC cells.
Results:
TRIM52-AS1 was revealed to be upregulated in HCC tissue samples according to GEPIA database. Consistent results were recognized in HCC cell lines. Subsequently, loss-of-function assays confirmed that TRIM52-AS1 ablation depressed cell proliferation, migration, invasion, and epithelial-to-mesenchymal transition process in vitro and inhibited tumor growth in vivo. Furthermore, the authors validated TRIM52-AS1 bound with miR-514a-5p in HCC. TRIM52-AS1 inversely regulated miR-514a-5p expression. Afterward, MRPS18A was identified to be a downstream target of miR-514a-5p. Ultimately, rescue assays manifested that MRPS18A upregulation could neutralize the attenuated effects resulting from TRIM52-AS1 deficiency.
Conclusions:
All in all, TRIM52-AS1 sponged miR-514a-5p to facilitate HCC progression through increasing MRPS18A expression. The findings highlight TRIM52-AS1 as a novel therapeutic target for HCC.
Introduction
Hepatocellular carcinoma (HCC), affecting 500,000 people per year, has become the sixth commonest malignant tumor and the third leading cause of death related to cancers all over the world. 1,2 High morbidity and mortality are chief characteristics of HCC. 3 Liver resection and transplantation are potential treatment methods for HCC and, to date, transarterial chemoembolization (TACE) is emerging as a new therapeutic method for HCC. It is a form of intraarterial catheter chemoembolization technique alternatively conveying cytotoxic drugs to the tumor bed. 4 Moreover, TACE is now regarded as an important part of the standard treatment for patients who are not suitable for surgical treatment. 5,6
However, the prognosis of HCC remains sadly poor and this is mainly owing to metastasis, recurrence, and diagnostic retardation. 7,8 Despite multiple reports about genes that participate in HCC, its underlying regulatory molecular mechanisms remain obscure. Therefore, to reveal novel molecular biomarkers and develop new therapeutic strategies for HCC, it is necessary to understand its pathophysiological mechanisms.
Recently, prominent attention has been paid to research of noncoding RNAs in the field of molecular biology. 9 Long noncoding RNAs (lncRNAs) are a class of noncoding RNA transcripts of more than 200 nucleotides in length without coding potential. 10 Increasing numbers studies have confirmed correlation of lncRNAs with diverse diseases, including cancer. For example, lncRNA GAPLINC contributes to colorectal cancer cell migration and invasion by modulating the miR-34a/c-MET Signal Pathway. 11 LncRNA AB073614 promotes cell proliferation and migration through regulating epithelial-to-mesenchymal transition (EMT) in glioma. 12 LncRNA NNT-AS1 aggravates lung cancer cell proliferation and invasion by sponging miR-129-5p. 13
Previously, TRIM52-AS1 was identified as playing a role of tumor promoter to regulate cell proliferation, migration, and apoptosis in renal cell carcinoma. 14 Although TRIM52-AS1 has been fully elucidated in renal cell carcinoma, the exact role and regulatory mechanism of TRIM52-AS1 in HCC remains ambiguous.
In this study, the authors executed experiments to study the expression pattern, the function, and underlying mechanism of TRIM52-AS1 in HCC. The results illustrate that TRIM52-AS1 sponged miR-514a-5p facilitating HCC progression by increasing mitochondrial ribosomal protein S18a (MRPS18A), which helps to develop novel therapeutic strategies for HCC treatment.
Materials and Methods
Cell lines and transfection plasmids
Human HCC cell lines (HepG2, SK-Hep-1, Huh7, and HCCLM3) and human normal liver cell line (L02) were all cultured in the dulbecco's modified eagle medium (Gibco, Grand Island, NY) under 5% CO2 and 37°C, procured from ATCC (Rockville, Maryland). One percent antibiotics and 10% fetal bovine serum (Gibco) acted as the supplements for culture medium. For transfection in HepG2 and Huh7 cells, TRIM52-AS1-specific shRNAs and control shRNAs (sh-NC), miR-514a-5p mimics and NC mimics, and pcDNA3.1/MRPS18A and empty pcDNA3.1 vector, all these plasmids were produced at GenePharma (Shanghai, China). Transfection Kit Lipofectamine 2000 was purchased from Invitrogen (Carlsbad, CA).
RNA isolation and real-time quantitative polymerase chain reaction
RNAs were initially isolated from HepG2 and Huh7 cell samples in line with the instruction of TRIzol from Invitrogen then converted to cDNA applying Reverse Transcription Kit (TaKaRa, Otsu, Japan). qPCR was conducted for quantification using Power SYBR Green (TaKaRa). Gene expression was calculated by the comparative delta-delta Ct method (2−ΔΔCt), relative to GAPDH or U6.
Cell Counting Kit-8 assay
Cells of Huh7 and HepG2 were transfected for 48 h and planted into the 96-well plates (4 × 103 cells/well) for incubation with Cell Counting Kit-8 (CCK-8) reagent (10 μL). The cell viability was monitored by testing the absorbance at 450 nm.
Colony formation
Transfected HCC cells were seeded at the density of 8 × 102 cells per well in the six-well plates for 2 weeks, following fixing and staining with 4% paraformaldehyde and 0.1% crystal violet. Clones were counted manually.
Flow cytometry for apoptosis
HCC cells were transfected and collected for resuspending in the 100 μL of binding buffer adding 2.5 μL of FITC-conjugated Annexin V and 1 μL of PI in the dark. Fifteen minutes later, the reaped cells were subjected to flow cytometry analysis (BD Biosciences, Franklin Lakes, NJ).
JC-1 analysis
Cells of HepG2 and Huh7 were collected after transfection for mixing with 10 mM of JC-1 Assay Kit (Beyotime, Shanghai, China). Thirty minutes later, the fluorescence labeled cells in PBS were analyzed with EnSpire Reader (PerkinElmer, Waltham, MA).
Transwell assays
The 24-well transwell inserts (Corning, New York, NY) with or without coating Matrigel (BD Biosciences, Bedford, MA) were acquired for invasion or migration. A total of 5 × 104 cells of HepG2 and Huh7 were seeded into the upper chamber for 24 h of incubation, with the complete growth medium in the lower chamber. Invading or migrating cells after fixation were then colored in 0.5% crystal violet and counted under microscope at 200 × magnification.
Western blot
The protein extracts from HCC cells were separated on 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), shifted onto polyvinylidene fluoride (PVDF) membranes, and then cultured with 5% nonfat milk. Primary antibodies against negative control GAPDH, Vimentin, N-cadherin, E-cadherin, CYP2S1, BCL2L13, ATP2C1, USP11, MRPS18A, and their appropriate HRP-tagged secondary antibodies were all procured from Abcam (Cambridge, MA). Band signals were monitored with gel imaging system.
Knockout of TRIM52-AS1 by CRISPR/Cas9
To obtain TRIM52-AS1 knockout (KO) Huh7 cells, a dual gRNA approach was applied in this study according to the procedure described in previous literatures. 15 In short, Huh7 cells cultured in a six-well plate were simultaneously transfected with dual gRNA and donor vector by use of DNA fectin (Applied Biological Materials). Seven days later, the transfected cells were selected using 1 μg/mL of puromycin treatment and then further incubated for about 2 weeks. Thereafter, colonies with puromycin resistance were picked out, and the KO clones were further identified by genomic PCR and qRT-PCR.
In vivo tumor growth experiments
Male BALB/c nude mice (6 weeks old) were procured from Experimental Animal Center of Chinese Academy of Sciences (Shanghai, China) and housed in pathogen-free conditions, with the approval from the Animal Care Committee of Xiangya Hospital, Central South University. One hundred microliters of parental Huh7 cells transfected with sh-NC or sh-TRIM52-AS1#1, as well as TRIM52-AS1 KO Huh7 cells transfected with pcDNA3.1 vectors containing TRIM52-AS1, were inoculated subcutaneously to mice, with tumor volume recorded every 4 d. Twenty-eight days postinjection, mice were sacrificed, and tumors were collected and weighed.
Subcellular fractionation
A total of 1 × 106 cells of HepG2 or Huh7 were harvested and suspended in the cell fraction buffer to collect cell cytoplasm and then cultivated in cell disruption buffer to collect cell nuclei. After RNA isolation, expression levels of TRIM52-AS1, GAPDH, and U6 were assessed by real-time quantitative polymerase chain reaction (RT-qPCR).
RNA pull down assay
The protein extracts from HCC cells were subjected to incubation with the biotinylated TRIM52-AS1 probes overnight, followed by further incubation with streptavidin agarose magnetic beads for 1 h. The retrieved miRNAs were analyzed by RT-qPCR.
RNA immunoprecipitation assay
A total of 1 × 107 HCC cells were reaped from RNA immunoprecipitation (RIP) lysis buffer and cultivated in the RIP buffer covering the beads-bound antibodies against human Ago2 and control IgG (Millipore, Bedford, MA). After digestion and RNA isolation, samples were assayed by RT-qPCR.
Dual-luciferase reporter assays
The wild-type (WT) and mutated (Mut) fragments of miR-514a-5p covering TRIM52-AS1 or MRPS18A binding sites were synthesized and cloned into Dual-Luciferase Vector pmirGLO (Promega, Madison, WI). The constructs TRIM52-AS1-WT/Mut and MRPS18A-WT/Mut were formed and cotransfected with miR-514a-5p mimics or NC mimics in HCC cells. After 48 h the samples were analyzed by Dual-Luciferase Reporter Assay System (Promega).
Statistical analyses
Each assay contained three or more bio-repeats, and results were displayed as the mean ± standard deviation. Significance analyses were implemented through Student's t-test and one-way analysis of variance (ANOVA) using Prism version 5.0 (GraphPad Software, La Jolla, CA), with p < 0.05 as threshold of statistical significance.
Results
TRIM52-AS1 was increased in HCC and TRIM52-AS1silence suppressed HCC progression
Previous studies reported that TRIM52-AS1 was involved in renal cell carcinoma, so the authors applied GEPIA database and found that TRIM52-AS1 expression was notably upregulated in LIHC (liver HCC) tissue samples (T = 369) compared with normal liver tissue samples (n = 160) (Fig. 1A). The finding encouraged us to further explore TRIM52-AS1 in HCC. RT-qPCR analysis disclosed that the expression of TRIM52-AS1 was increased in HCC cell lines relative to normal liver cell L02 (Fig. 1B). The authors observed that HepG2 and Huh7 exhibited higher expression of TRIM52-AS1, so these two cells were used for further study.
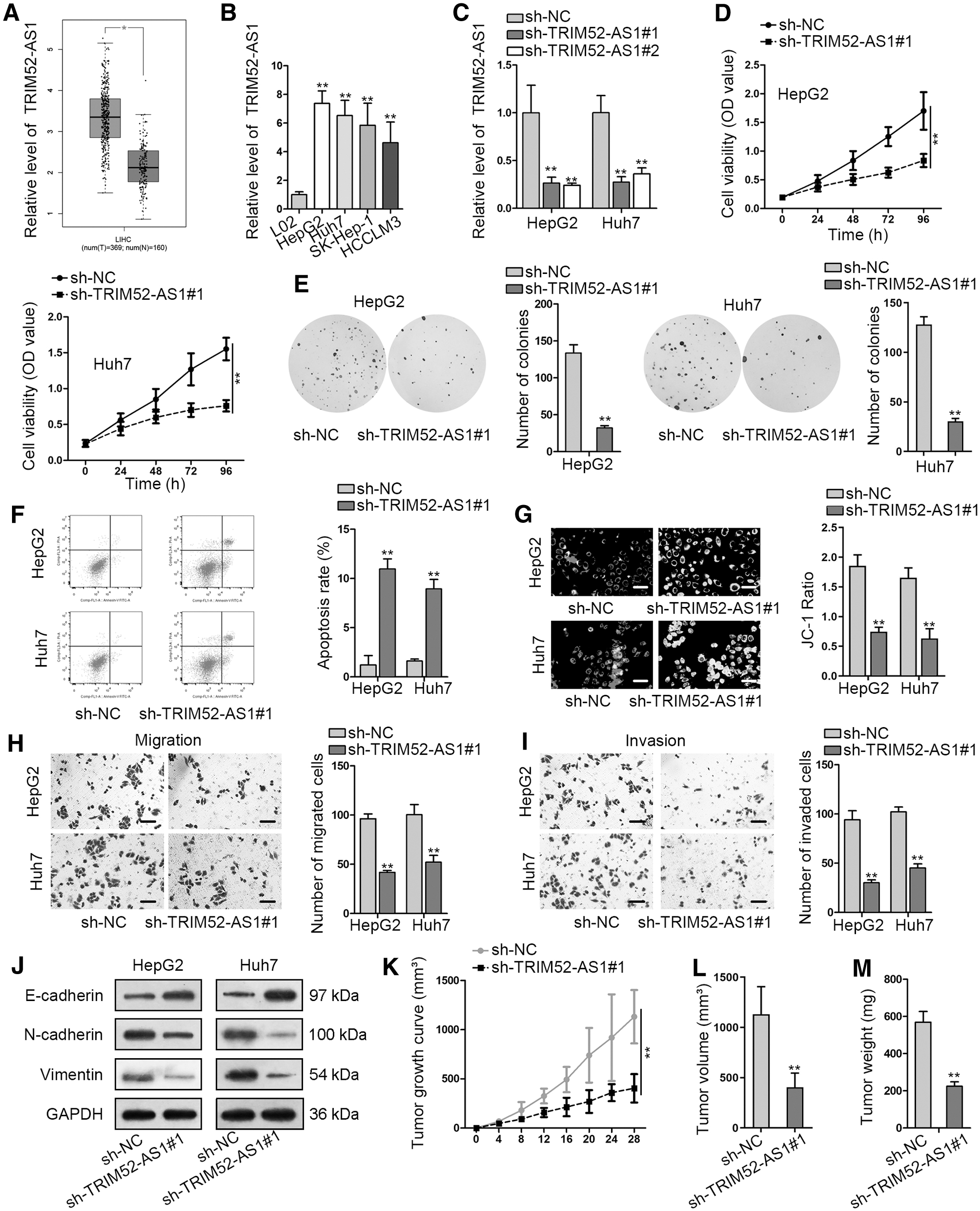
TRIM52-AS1 was increased in HCC, and TRIM52-AS1silence suppressed HCC progression.
Thereafter, the data from RT-qPCR manifested that by transfection with sh-TRIM52-AS1#1 and sh-TRIM52-AS1#2 vectors, both sh-RNAs targeting TRIM52-AS1 reduced the expression of TRIM52-AS1 in HCC cells (Fig. 1C). The authors selected sh-TRIM52-AS1#1 for further use as it presented better knockdown efficiency. Later on, the authors analyzed the function of TRIM52-AS1 in HCC. As shown in CCK-8 assay, cell viability was inhibited by TRIM52-AS1 downregulation (Fig. 1D). Consistently, colony formation assay verified that cell proliferation was attenuated by reduction of TRIM52-AS1 (Fig. 1E). Next, cell apoptosis in HCC cells was assessed by flow cytometry analysis and JC-1 assays.
Data from flow cytometry analysis unveiled that cell apoptosis was boosted by inhibition of TRIM52-AS1 in HCC cells (Fig. 1F). JC-1 assay showed that mitochondrial membrane potential was significantly lessened by TRIM52-AS1 blockage (Fig. 1G). Transwell assays demonstrated that both cell migration and invasion were alleviated as a result of TRIM52-AS1 depletion (Fig. 1H, I). EMT process is an important marker of cancer metastasis, so that the authors detected the protein level of EMT-related proteins as well.
According to the results, the protein level of E-cadherin was overtly enhanced, whereas the protein level of Vimentin and N-cadherin was obviously cut down in sh-TRIM52-AS1#1 group in comparison with sh-NC group (Fig. 1J). Finally, in vivo xenograft assays were performed, and the authors found that silencing TRIM52-AS1 was able to block tumor growth of HCC in vivo, as the tumor volume and tumor weight were both attenuated by TRIM52-AS1 descent (Fig. 1K–M). Moreover, the authors proved that the in vivo growth of Huh7 cells was markedly blocked under TRIM52-AS1 KO, and such tumor suppression caused by TRIM52-AS1 KO was offset in response to reexpression of TRIM52-AS1 (Supplementary Fig. S1). These results further intensified the significance of TRIM52-AS1 in accelerating HCC tumorigenesis.
To conclude, TRIM52-AS1 expression was increased in HCC, and TRIM52-AS1 silence suppressed HCC progression.
TRIM52-AS1 bound with miR-514a-5p in HCC
The function of TRIM52-AS1 in HCC was elucidated above; the authors next investigated the regulatory mechanism of TRIM52-AS1 in HCC. Nuclear–cytoplasmic fractionation revealed that TRIM52-AS1 was mainly in cytoplasm (Fig. 2A), suggesting that TRIM52-AS1 might bind with miRNAs in HCC. The authors searched star-Base and found that there were 20 miRNAs with potential binding to TRIM52-AS1 (Fig. 2B). Of the 20, the role of 13 miRNAs in HCC has not been investigated. Moreover, RNA pull down assay unveiled that only miR-514a-5p was greatly enriched in bio-TRIM52-AS1-WT group rather than in bio-NC and bio-TRIM52-AS1-Mut groups (Fig. 2C). RT-qPCR detected the expression of miR-514a-5p, disclosing that miR-514a-5p was downregulated in HCC cells (Fig. 2D).

TRIM52-AS1 bound with miR-514a-5p in HCC.
In addition, miR-514a-5p expression was inversely regulated by TRIM52-AS1 (Fig. 2E). RIP manifested the enrichment of TRIM52-AS1, and miR-514a-5p was observed in Ago2 group instead of IgG group (Fig. 2F). Then RT-qPCR obtained the satisfactory overexpression efficacy of miR-514a-5p in HCC cells (Fig. 2G). The putative binding site for TRIM52-AS1 and miR-514a-5p was presented in Figure 2H. Luciferase reporter assay showed that miR-514a-5p upregulation reduced the luciferase activity of TRIM52-AS1-WT, but didn't influence that of TRIM52-AS1-Mut (Fig. 2I). All the data indicated that TRIM52-AS1 bound with miR-514a-5p.
MRPS18A functioned as a downstream target of miR-514a-5p
Next, the authors analyzed the downstream of miR-514a-5p. Two programs (including miRmap and RNA22) selected from starBase revealed that CYP2S1, BCL2L13, ATP2C1, USP11, and MRPS18A were potentially targeted by miR-514a-5p (Fig. 3A, B). RT-qPCR disclosed that miR-514a-5p overexpression markedly decreased the mRNA and protein expression of MRPS18A, whereas had no obvious effects on the expression of others in both HepG2 and Huh7 cells (Fig. 3C, D). In addition, MRPS18A expression was evidently enhanced in HCC cells in comparison with normal liver cells (Fig. 3E).

MRPS18A functioned as a downstream target of miR-514a-5p.
In addition, TRIM52-AS1, miR-518a-5p, and MRPS18A were identified to be enriched in Ago2-precipitated complexes (Fig. 3F), suggesting that miR-518a-5p might bind with MRPS18A in RNA-induced silencing complexes in HCC. The binding sites between miR-518a-5p and MRPS18A from starBase are shown in Figure 3G. Luciferase reporter assays further confirmed that miR-518a-5p could bind with and silence MRPS18A, proved by overexpression of miR-518a-5p that markedly reduced the luciferase activity of MRPS18A-WT, not that of MRPS18A-Mut (Fig. 3H). In summary, MRPS18A served as a downstream target of miR-514a-5p in HCC.
TRIM52-AS1 accelerated HCC progression by regulating MRPS18A
Finally, the authors performed rescue assays to validate whether TRIM52-AS1 accelerated HCC progression by regulating MRPS18A. MRPS18A was overexpressed in HepG2 cells by pcDNA3.1/MRPS18A, which resulted in a marked enhancement of MRPS18A expression (Fig. 4A). CCK-8 and colony formation assays uncovered that MRPS18A upregulation recovered TRIM52-AS1-mediated suppression on cell proliferation in HCC (Fig. 4B, C). Additionally, enhanced cell apoptosis by TRIM52-AS1 silence was reversed by MRPS18A overexpression (Fig. 4D, E).

TRIM52-AS1 accelerated HCC development by regulating MRPS18A.
Transwell assays revealed that cell migration and invasion repressed by TRIM52-AS1 depletion was rescued as a result of MRPS18A enhancement (Fig. 4F, G). Western blot analysis manifested that overexpression of MRPS18A neutralized the promoting effect on E-cadherin, as well as the inhibitory effects on N-cadherin and Vimentin caused by TRIM52-AS1 ablation (Fig. 4H). Altogether, TRIM52-AS1 accelerated HCC development through modulating MRPS18A.
Discussion
Despite being one of the most frequently diagnosed cancers, HCC is still associated with poor clinical outcome. 16,17 Therefore, novel techniques for HCC treatment are urgently called for. It has been widely accepted that lncRNAs are implicated in HCC progression. For instance, lncRNA CPS1-IT1 blocks HCC metastasis by modulating HIF-1α activity and inactivating EMT. 18 LncRNA PARP1 increases the expression of PARP1 to drive the progression of HCC. 19 LncRNA GAS5 hinders HCC metastasis although binding with miR-182 to regulate ANGPTL1 expression. 20
Nonetheless, the role and function of TRIM52-AS1 in HCC remains unclear. Based on the data from GEPIA database, TRIM52-AS1 expression was evidently upregulated in HCC tissue samples. Their experiment data also confirmed TRIM52-AS1 expression elevation in HCC cell lines. Functionally, TRIM52-AS1 attenuation markedly impeded cell proliferation, migration, and invasion, as well as EMT process in vitro. Meanwhile, its knockdown or KO led to hindered HCC tumor growth in vivo. On the whole, this study unveiled for the first time that TRIM52-AS1 served an oncogenic role to drive HCC progression.
Competing endogenous RNAs (ceRNAs) are extensively researched as novel biomarkers for cancer diagnosis and prognosis. Downregulation of lncRNA ANRIL impairs head and neck squamous cell carcinoma tumorigenesis by regulating FGFR1 expression by binding with miR-125a-3p. 21 LIN28B mediated-lncRNA NEAT1 accelerated high-grade serous ovarian cancer cell proliferation and migration through sponging miR-506. 22 LncRNA GACAT3 sponges miR-497 and regulates CCND2 to contribute to breast cancer cell proliferation. 23
However, whether TRIM52-AS1 can function as a ceRNA in HCC needs to be further explored. In present study, bioinformatics analysis indicated that miR-514a-5p had high potentials to bind with TRIM52-AS1. MiR-514 was verified to suppress cell proliferation and enhance chemoresistance in ovarian cancer through regulating ATP binding cassette subfamily. 24 But the role of miR-514a-5p in HCC has not been reported. In this study, TRIM52-AS1 was corroborated to bind with miR-514a-5p in HCC cells. MiR-514a-5p was negatively regulated by TRIM52-AS1. Taken together, TRIM52-AS1 sponged miR-514a-5p to facilitate HCC progression.
MRPS18A expression was recognized to be upregulated in breast cancer. 25 In present study, MRPS18A was selected as the downstream of TRIM52-AS1/miR-514a-5p axis in HCC. Moreover, the authors validated that MRPS18A expression was increased in HCC cells, and MRPS18A upregulation restored the inhibitory effects of TRIM52-AS1 downregulation on the biological functions of HCC cells.
All in all, this work proved that TRIM52-AS1 sponged miR-514a-5p to facilitate HCC progression by increasing MRPS18A, which proposed possible directions for exploring possible diagnostic biomarkers or therapeutic targets for HCC.
Footnotes
Acknowledgment
The authors appreciate the technical supports of laboratory members.
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.