
Abstract
Background:
Pancreatic cancer (PC) is a leading cause of cancer-related deaths worldwide. Human leukocyte antigen complex P5 (HCP5), a member of long noncoding RNAs (lncRNAs), was reported to be associated with the poor prognosis of PC. However, the mechanism of HCP5 in regulating the progression of PC remains poorly defined.
Materials and Methods:
Quantitative real-time polymerase chain reaction was performed to detect the expression levels of HCP5, microRNA (miR)-140-5p, and cyclin-dependent kinase 8 (CDK8) in PC tissues and cells. Cell counting kit-8 (CCK-8) assay was utilized to check cell proliferation. Transwell assay was employed to evaluate the abilities of cell migration and invasion. Xenograft tumor model was established to investigate the biological role of HCP5 in PC in vivo. The interaction between miR-140-5p and HCP5 or CDK8 was predicted by starBase or TargetScan, respectively. The dual-luciferase reporter assay was conducted to corroborate the interaction. The protein level of CDK8 was measured by Western blot.
Results:
HCP5 and CDK8 were significantly upregulated in PC tissues and cells, opposite to the expression of miR-140-5p. High expression of HCP5 contributed to the low survival rate and HCP5 silencing inhibited proliferation, migration, and invasion of PC cells in vitro. Simultaneously, in vivo experiments indicated that downregulation of HCP5 suppressed tumor growth. In addition, miR-140-5p was a target of HCP5 and bound to the 3′-untranslated region (3′UTR) of CDK8. Further studies revealed that overexpression of CDK8 reversed the miR-140-5p-mediated inhibitory effect on PC progression. Moreover, downregulation of miR-140-5p or upregulation of CDK8 inverted the silencing-mediated repressive impact of HCP5 on PC progression.
Conclusion:
Downregulation of HCP5 impeded PC progression by downregulating CDK8 via sponging miR-140-5p.
Introduction
Pancreatic cancer (PC) is an increasing threat to humans and leads to many deaths globally. 1 Early PC shows little symptoms and the disease is usually advanced and spreads to other parts of the body when the diagnosis is made. 2,3 Hence, it is imperative to find novel therapeutic methods and molecular targets for PC treatment.
Long noncoding RNAs (LncRNAs) are a class of RNA molecules (>200 nucleotides) and cannot encode proteins. 4 LncRNAs are verified to partake in the regulation of the progression of various human cancers. 5 –7 Recent reports have reported that lncRNA human leukocyte antigen complex P5 (HCP5) is dysregulated in cutaneous melanoma, 8 breast cancer, 9 and lung adenocarcinoma. 10 Wang et al. found that HCP5 was correlated with the poor prognosis of PC. 11 However, the role of HCP5 in modulating PC progression has not been fully addressed.
MicroRNAs (miRNAs) are small RNA molecules (probably 22 nucleotides) and mediate gene expression by guiding Argonaute proteins to target sites in the 3′-untranslated region (3′UTR) of messenger RNA (mRNA). 12 Growing evidence has shed light on the fact that miRNAs play a crucial part in various human cancers. 13,14 Liang et al. confirmed that miR-140-5p was involved in the modulation of PC progression. 15 Nevertheless, the regulatory mechanism of miR-140-5p in PC needs to be further explored.
Cyclin-dependent kinase 8 (CDK8) functions in diverse human diseases. Guo et al. found that inhibition of CDK8 activity repressed autoimmune diseases. 16 Song et al. demonstrated that CDK8 regulated the chemosensitivity of breast cancer cells to trastuzumab. 17 A recent article showed that CDK8 modulated the angiogenesis of PC cells. 18 Therefore, CDK8 may be an attractive drug target for PC and new regulators regulating CDK8 require to be determined.
In this study, the expression level of HCP5 in PC tissues and cells was checked. The function and potential regulatory mechanism of HCP5 in PC were further studied by subsequent experiments.
Materials and Methods
Samples and cell culture
Forty-eight PC tissues and paired nearby noncancerous tissues were collected from Dongying City People's Hospital. Informed consent was obtained from every patient and our research was authorized by the Ethics Committee of Dongying City People's Hospital. The human pancreatic duct epithelial cell line (HPDE6-C7) was purchased from Bogoo Biotech (Shanghai, China) and PC cell lines (PANC-1, BxPC-3, SW1990, and ASPC-1) were obtained from the American Type Culture Collection (Manassas, VA). McCoy's 5A medium (Sigma, St. Louis, MO), within 5% CO2 and 10% fetal bovine serum (FBS; Sigma) was utilized to culture cells.
Cell transfection
Small interfering RNA against HCP5 (named as si-HCP5), miR-140-5p mimic (named as miR-140-5p), miR-140-5p inhibitor (named as anti-miR-140-5p), and corresponding controls (si-NC, miR-NC, and anti-miR-NC) were obtained from GenePharma (Shanghai, China). CDK8 overexpression plasmid (named as CDK8) and its control (pcDNA) were acquired from RiboBio (Guangzhou, China). Cell transfection was carried out by Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) referring to the given procedures.
RNA isolation and quantitative real-time polymerase chain reaction
Total RNA from PC tissues and cells was extracted using the TRIzol reagent (Vazyme, Nanjing, China), which was then reversely transcribed to complementary DNA (cDNA) by PrimeScript™ RT Master Mix kit (Takara, Dalian, China). Afterward, quantitative real-time polymerase chain reaction (qRT-PCR) was conducted by SYBR Green PCR Master Mix (Vazyme) and data were analyzed using 2−ΔΔCt method. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 were introduced as the inner controls. Primers in this study:
HCP5 (Forward 5′-CCGCTGGTCTCTGGACACATACT-3′, reverse 5′-CTCACCTGTCGTGGGATTTTGC-3′); miR-140-5p (forward, 5′-ACACTCCAGCTGGGAGGCGGGGCGCCGCGGGA-3′, reverse 5′-CTCAACTGGTGTCGTGGA-3′); CDK8 (forward, 5′-GCCGGTTGTCAAATCCCTTAC-3′, reverse 5′-TGTGACTGCTGTCTTGATTCCCT-3′); GAPDH (forward 5′-CCACTCCTCCACCTTTGAC-3′, reverse, 5′-ACCCTGTTGCTGTAGCCA-3′); U6 (forward, 5′-TGCGGGTGCTCGCTTCGGCAGC-3′, reverse, 5′-CCAGTGCAGGGTCCGAGGT-3′).
Cell counting kit-8 assay
After transfection, ASPC-1 and SW1990 cells were seeded into 96-well plates and then incubated with 10 μL cell counting kit-8 (CCK-8) solution (Beyotime, Shanghai, China) for 2 h. Optical density values were examined at 450 nm wavelength under the microplate reader (Bio-Rad, Richmond, Virginia).
Transwell assay
Transwell chamber precoated with Matrigel (Beyotime) or not was utilized to check the cell invasion or migration, respectively. Transfected cells were seeded into the upper chamber and medium containing FBS was placed in the lower chamber. After being treated with crystal violet staining solution (Beyotime), migrated or invaded cells were analyzed using an inverted microscope (MTX Lab Systems, Bradenton, FL).
Xenograft mice model
Five-week-old nude mice were acquired from LingChang Biotech (Shanghai, China). Lentivirus harboring short hairpin RNA targeting HCP5 (sh-HCP5) and negative control (sh-NC) were constructed by GeneCopoeia (Rockville, MD, USA). Subsequently, ASPC-1 cells infected with sh-HCP5 or sh-NC were implanted subcutaneously into the flank of the mice. The tumor volume was calculated every 3 d according to the formula: 0.5 × length × width 2 . The tumor weight was calculated after the mice were euthanized and the level of HCP5 in tumors was checked by qRT-PCR. The animal experiment was authorized by the Animal Care and Use Committee of Dongying City People's Hospital and performed following the instructions of the animal protection and ethics institute.
Dual-luciferase reporter assay
The possible complementary sequences between miR-140-5p and HCP5 or CDK8 were forecasted by starBase 19 or TargetScan, 20 respectively. The wild-type sequence of HCP5 or CDK8 3′UTR harboring the binding sites of miR-140-5p was inserted into the pGL3 vector (Promega, Madison, WI) to generate reporter vector HCP5-WT or CDK8-WT, respectively. Similarly, HCP5-MUT and CDK8-MUT reporter vectors were established by mutating the forecasted target sequences of miR-140-5p. Subsequently, miR-140-5p or its control with these vectors was cotransfected into ASPC-1 and SW1990 cells using Lipofectamine 2000 (Invitrogen). The Dual-Luciferase Assay kit (Vazyme) was employed to check luciferase activity.
Western blot
Proteins from samples were isolated using RIPA buffer (Vazyme) and corresponding concentrations were measured by Bradford Protein Quantification Kit (Beyotime). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto the nitrocellulose membrane (Beyotime). The membranes were blocked with 5% skimmed milk (Beyotime) and then were incubated with the primary antibodies: anti-CDK8 (1:5000, SC-1521; Santa cruz, Dallas, TX) or GAPDH (1:2500, ab9485; Abcam, Cambridge, United Kingdom) overnight. Subsequently, the membranes were incubated with the secondary antibody (1:3000, ab205718; Abcam) for 3 h. The membranes were analyzed by the ChemiDoc™ MP Imaging System (Bio-Rad) after being treated with ECL kit (Beyotime).
Statistical analysis
Experimental data were calculated by GraphPad Prism (GraphPad, La Jolla, CA) and presented by mean ± standard deviation. Student's t-test was utilized to assess the difference of two independent groups. For more than two groups, the one-way analysis of variance was utilized to check the difference. Pearson's correlation coefficient was applied to analyze the correlation among HCP5, miR-140-5p, and CDK8 in PC tissues. Survival curves were plotted using Kaplan–Meier methodology with log-rank test. Every experiment was repeated at least three times independently. p < 0.05 represented statistical significance.
Results
HCP5 was strikingly upregulated in PC and correlated with the poor prognosis
To investigate the role of HCP5 in PC, its expression level was measured and the results showed HCP5 was notably upregulated in PC tissues and cell lines (PANC-1, BxPC-3, SW1990, and ASPC-1) compared with corresponding normal tissues and cell line (HPDE6-C7) (Fig. 1A, B). Afterward, overall survival rate was calculated and the data indicated that the higher level of HCP5 resulted in the lower survival rate (Fig. 1C). The data of clinicopathologic features of patients showed that HCP5 was significantly correlated with TNM stage, tumor size, lymph-node metastasis, and tumor differentiation (Table 1). These results supported the view that HCP5 might act as an oncogenic role in PC progression.
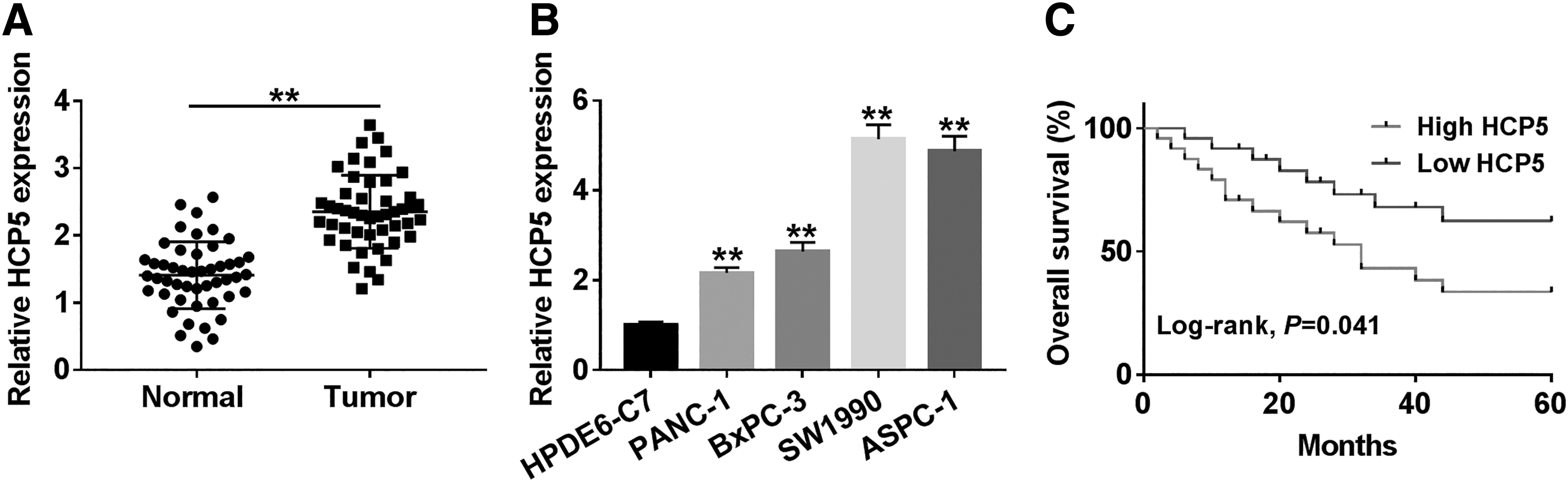
HCP5 was highly expressed in PC tissues and cells.
Clinicopathological Factors and HCP5 Expression in Patients with Pancreatic Cancer
p < 0.05.
Chi-square test.
HCP5, human leukocyte antigen complex P5.
Downregulation of HCP5 inhibited cell proliferation, migration, invasion, and tumor growth
To explore the function of HCP5 in PC, ASPC-1 and SW1990 cells were first infected with si-HCP5 lines (si-HCP5#1, si-HCP5#2, and si-HCP5#3) and the knockdown efficiency was confirmed (Fig. 2A). Thereafter, cell proliferation was measured and the data indicated that knockdown of HCP5 markedly hindered proliferation of ASPC-1 and SW1990 cells (Fig. 2B, C). Transwell assay showed that HCP5 silencing conspicuously weakened the abilities of migration and invasion of PC cells (Fig. 2D, E). To confirm the biological function of HCP5 in PC in vivo, we established the xenograft mouse model using ASPC-1 cells transfected with sh-NC or sh-HCP5. The results showed that HCP5 depletion contributed to the clear shrink in tumor volume (Fig. 2F) and decline in tumor weight (Fig. 2G). Besides, the level of HCP5 was clearly decreased in sh-HCP5 group (Fig. 2H). Moreover, we found that upregulation of HCP5 significantly enhanced cell proliferation, migration, and invasion in both ASPC-1 and SW1990 cells (Supplementary Fig. S1). Collectively, these results illustrated that HCP5 promoted PC progression in vitro and in vivo.

Knockdown of HCP5 blocked proliferation, migration, and invasion of PC cells and tumor growth.
HCP5 targeted miR-140-5p and negatively regulated the expression of miR-140-5p in PC
Many studies have reported that lncRNAs could target miRNAs to regulate the progression of cancers. 6,21 In this research, miR-140-5p was predicted to be a target of HCP5 by starBase (Fig. 3A). Also, the dual-luciferase reporter assay indicated that miR-140-5p efficiently reduced the luciferase activity of HCP5-WT reporter vector but not that of HCP5-MUT reporter vector (Fig. 3B, C). Further studies showed that overexpression of HCP5 significantly downregulated miR-140-5p in PC cells, while knockdown of HCP5 apparently upregulated miR-140-5p (Fig. 3D, E). Also, miR-140-5p was clearly declined in PC tissues (Fig. 3F). Moreover, the expression of miR-140-5p was negatively associated with HCP5 in PC tissues (Fig. 3G). Altogether, these results illuminated that HCP5 interacted with miR-140-5p and downregulated miR-140-5p in PC tissues and cells.
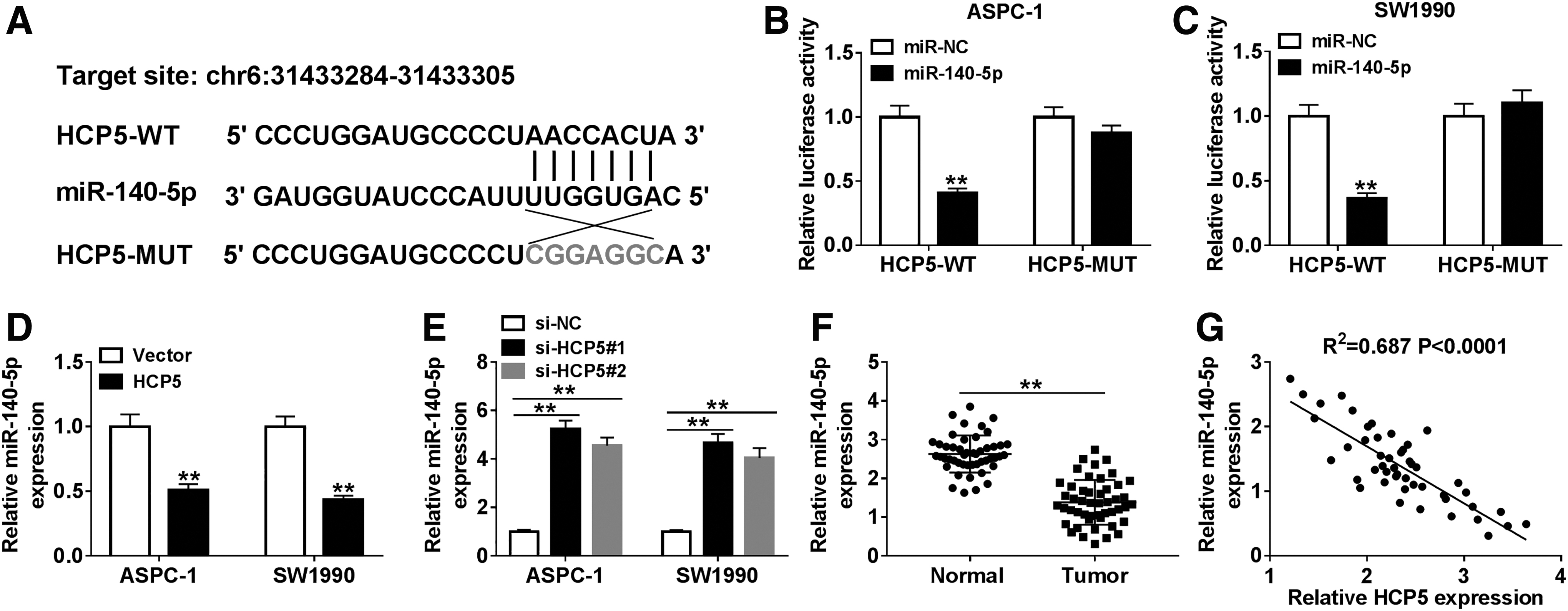
HCP5 interacted with miR-140-5p and negatively regulated miR-140-5p in PC tissues and cells.
CDK8 was a target of miR-140-5p and negatively modulated by miR-140-5p
To further investigate the regulatory mechanism of miR-140-5p, it is possible that target genes were predicted by TargetScan and the result showed that miR-140-5p could bind to the 3′UTR of CDK8 (Fig. 4A), which was confirmed by the dual-luciferase reporter assay (Fig. 4B, C). Meanwhile, the mRNA level of CDK8 was obviously declined in miR-140-5p group, whereas CDK8 was strikingly upregulated in anti-miR-140-5p group (Fig. 4D, E). Also, miR-140-5p mimic evidently decreased the protein level of CDK8 in PC cells, while miR-140-5p inhibitor notably increased the level of CDK8 (Fig. 4F, G), respectively. In addition, CDK8 was significantly upregulated in PC tissues (Fig. 4H) and its expression was negatively correlated with miR-140-5p (Fig. 4I). To sum up, these results testified that miR-140-5p targeted CDK8 and negatively regulated the expression of CDK8 in PC.

MiR-140-5p bound to the 3′UTR of CDK8 and negatively regulated CDK8 in PC tissues and cells.
Overexpression of CDK8 reversed miR-140-5p-mediated repressive effects on PC progression
To probe the regulatory relationship between CDK8 and miR-140-5p in PC progression, the mRNA and protein levels of CDK8 in PC cells infected with miR-140-5p, CDK8, or miR-140-5p+CDK8, in addition to matched controls were first detected. The result showed that CDK8 was markedly downregulated under miR-140-5p mimic treatment, whereas its expression was obviously elevated following the infection with CDK8 (Fig. 5A–C). Meanwhile, CCK-8 assay indicated that overexpression of CDK8 significantly inhibited miR-140-5p-mediated repressive impact on proliferation of PC cells (Fig. 5D, E). Analogously, the inhibitory effects of miR-140-5p-mediated on migration and invasion were overturned by upregulating CDK8 (Fig. 5F, G). From these results, it could be concluded that miR-140-5p and CDK8 played opposite roles in PC progression, and miR-140-5p-mediated inhibitory effects on PC progression were abolished by CDK8 overexpression.
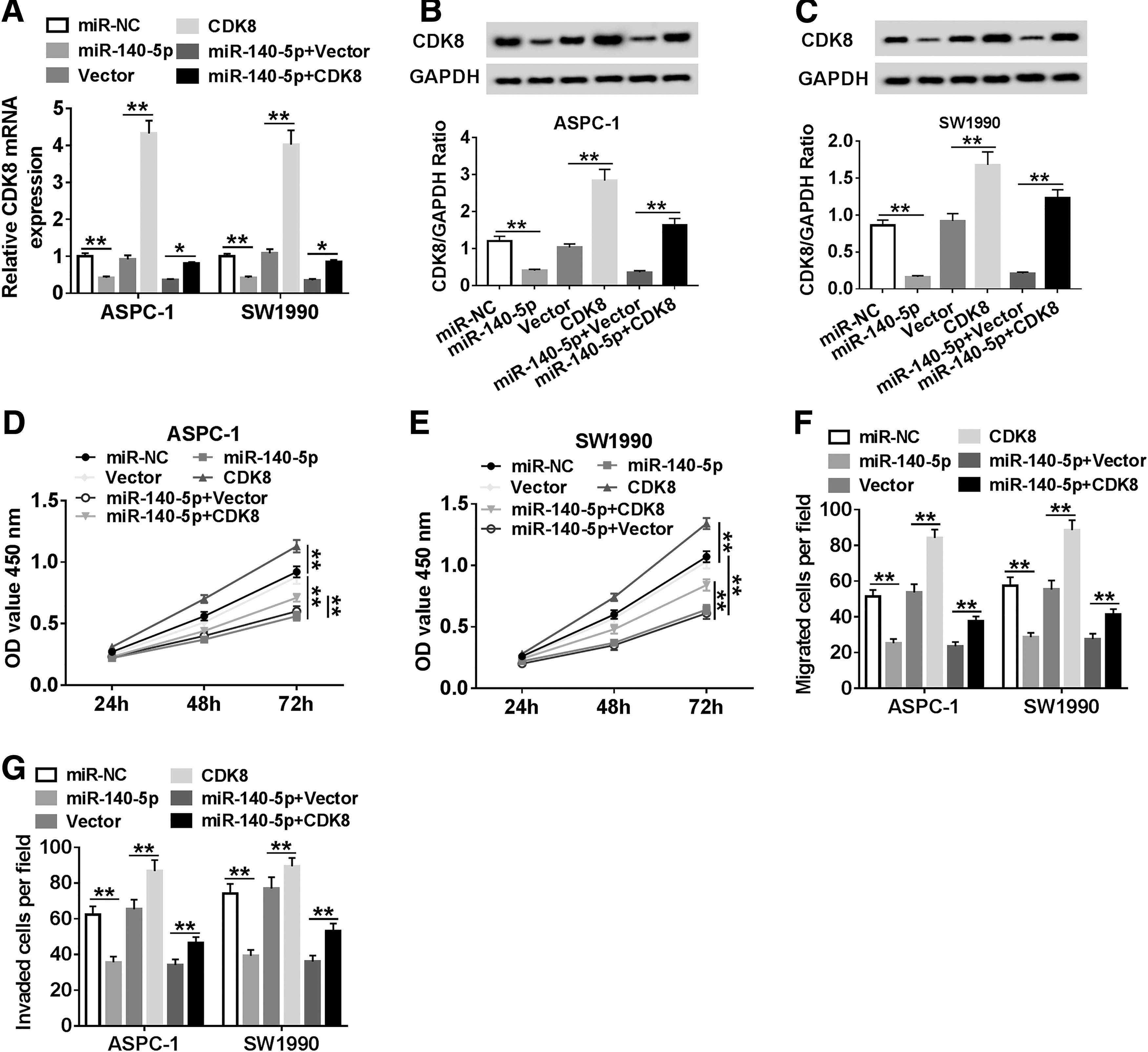
Enforced expression of CDK8 abolished miR-140-5p-mediated effects on proliferation, migration, and invasion of PC cells.
HCP5 regulated proliferation, migration, and invasion of PC cells by miR-140-5p/CDK8 axis
To elucidate out the underlying molecular mechanism of HCP5 in PC progression, the correlation between CDK8 and HCP5 was analyzed and the data showed that the expression of CDK8 was positively associated with HCP5 in PC tissues (Fig. 6A). Afterward, the mRNA and protein levels of CDK8 in PC cells infected with si-HCP5#1, si-HCP5#1+anti-miR-140-5p, and si-HCP5#1+CDK8, in addition to corresponding controls were detected. The results illustrated that the expression of CDK8 was conspicuously decreased in si-HCP5#1 group but prominently increased after the additional infection with anti-miR-140-5p or CDK8 (Fig. 6B–D). Further research revealed that miR-140-5p depletion or CDK8 overexpression transposed HCP5 silencing-mediated suppressive impact on proliferation of PC cells (Fig. 6E, F), and migration and invasion (Fig. 6G, H). Taken together, these results suggested that HCP5 mediated the progression of PC through miR-140-5p/CDK8 axis.

HCP5/miR-140-5p/CDK8 axis mediated PC progression.
Discussion
PC is the fourth dominant cause of cancer-related deaths in the United States and contributes to about 227,000 deaths every year worldwide. 22 Former research showed that lncRNA HCP5 functioned in the development of human cancers. Wei et al. reported that HCP5 suppressed cutaneous melanoma progression by interacting with miR-12. 8 Wang et al. found that HCP5 promoted triple negative breast cancer progression. 9 Jiang et al. confirmed that HCP5 promoted lung adenocarcinoma metastasis by miR-203/SNAI axis. 10 In this study, we checked the expression of HCP5 and the result showed that HCP5 was significantly upregulated in PC tissues and cells. Moreover, the high level of HCP5 contributed to the low survival rate. To study the function of HCP5 in PC progression, the proliferation, migration, and invasion of PC cells transfected with si-HCP5 were checked. The data clarified that knockdown of HCP5 notably repressed proliferation, migration, and invasion of PC cells in vitro. In addition, animal experiments indicated that HCP5 silencing inhibited tumor growth. These results demonstrated that HCP5 might function as an oncogene in PC progression.
LncRNA could regulate the progression of cancer by altering the expression of miRNA. 21 In our research, miR-140-5p was predicted to be a target of HCP5 and the interaction was verified. Afterward, the regulatory relationship of the two in PC was investigated and the data manifested that the level of miR-140-5p was negatively associated with HCP5. Besides, miR-140-5p was notably decreased in PC tissues, which was in line with Liu's report. 15 To further understand the regulatory mechanism of miR-140-5p in PC, its target gene, CDK8, was found and confirmed. CDK8 was reported to be correlated with various human cancers. 17,23 In this study, CDK8 was significantly increased in PC tissues, which was supported by a previous study. 18 Moreover, CDK8 was negatively regulated by miR-140-5p in PC cells in vitro. In-depth studies showed that miR-140-5p markedly restrained proliferation, migration, and invasion of PC cells, while overexpression of CDK8 rescued these effects. Since miR-140-5p was a target of lncRNA HCP5 and bound to the 3′UTR of CDK8, we further explored the potential mechanism among them. Our results indicated that the declined mRNA and protein levels of CDK8 in si-HCP5#1 group were clearly inverted following the infection with miR-140-5p inhibitor or CDK8. Simultaneously, HCP5 silencing-mediated inhibitory impacts on proliferation, migration, and invasion of PC cells were transposed by the downregulation of miR-140-5p or upregulation of CDK8. Taken together, our results suggested that lncRNA HCP5 acted as a sponge to interact with miR-140-5p and downregulate miR-140-5p, thus regulating the expression of CDK8, ultimately affecting the progression of PC. As a more specific mechanism has yet to be explained, we will continue to explore the downstream molecular mechanism of the HCP5/miR-140-5p/CDK8 axis in the subsequent study.
Conclusions
In conclusion, our research demonstrated that HCP5 and CDK8 were upregulated, while miR-140-5p was downregulated in PC. HCP5 silencing suppressed the progression of PC and HCP5 could sponge miR-140-5p to regulate CDK8 expression. Moreover, we found that HCP5 downregulation inhibited PC progression by reducing the expression of CDK8 via sponging miR-140-5p. Our findings indicated that HCP5 played a promotion role in PC and the HCP5/miR-140-5p/CDK8 axis might provide potential targets for improving the treatment of PC.
Footnotes
Authors' Contributions
Conception and design: B.Y., Q.G., and J.L.; Development of methodology: T.Y.; Acquisition of data: X.Z.; Analysis and interpretation of data: W.X.; Writing, review, and revision of article: B.Y., Q.G., and J.L.; All coauthors have reviewed and approved of the article before submission.
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.