
Abstract
Aim:
Hepatocellular carcinoma (HCC), as one primary liver cancer type, accounts for 75%–85% of liver cancer cases. HCC is the second leading cause of cancer death in East Asia and sub-Saharan Africa and the sixth most common in Western countries. Identification of key genes would facilitate the development of therapies and improve the prognosis outcomes of HCC patients. This study was to identify the key biological processes, pathways, and key genes in HCC.
Methods:
Data were downloaded from Broad GDAC. Differentially expressed genes (DEGs) and weighted gene coexpression network (WGCNA) were analyzed by DESeq2 and WGCNA, respectively. Gene ontology (GO) and KEGG enrichment analyses were performed on all DEGs and the coexpressed genes in two significant modules. Kaplan–Meier plotter online database was used to identify the potential prognostic genes in HCC. Finally, GEO database was used to validate the analysis of gene expression of Broad GDAC data.
Results:
The authors identified the dark gray and red modules as the significant modules in HCC based on WGCNA. GO and KEGG enrichment of the two significant modules identified the mitochondrion-mediated metabolic processes and pathways, and the cell cycle as the key biological processes and pathways in HCC. Subsequently, 28 hub genes were screened out by constructing protein–protein interaction network using Metascape. Finally, three genes (NDUFAF6, CKAP5, and DSN1 genes) were identified to be potential prognostic and key genes in HCC based on Kaplan–Meier survival analysis, GEO dataset validation, and literature review.
Conclusions:
The authors found that mitochondrion-mediated metabolic processes and the cell cycle were the key biological processes and pathways in HCC. NDUFAF6, CKAP5, and DSN1 genes were valuable genes with the potential to be prognosis biomarkers and targeted therapies in HCC.
Introduction
Hepatocellular carcinoma (HCC) has been linked to the death of millions of people with liver problems worldwide. It is the second leading cause of mortality in East Asia and sub-Saharan Africa, and the sixth most prevalent malignancy in many Western nations. 1,2 According to the 2018 global cancer statistics, HCC accounts for 75%–85% of liver cancer cases. 1 Hence, more humanitarian commitments are needed as the causes of this malignant disease include multiple factors such as chronic viral hepatitis, hepatitis virus, metabolic disease, alcohol consumption, and smoking. 3 –7
Currently, liver transplantation or surgical resection is the best therapeutic option to treat HCC, but the 5-year survival rate of HCC is merely 30%–40% after treatment. 8,9 This low survival rate is in part due to aberrant hepatocyte proliferation, oxidative stress, mitochondrial damage, and induction of reactive oxygen species, all of which are key biological HCC processes. 10 To increase the treatment efficacy of HCC, a comprehensive understanding of molecular and functional characterization is required. 11 The good news, however, is that the effective identification of HCC prognostic biomarkers is possible with the development of bioinformatics such as NUPR1 12 and DKK1 13 and with the advancement in gene identification and gene analysis tools.
For instance, Weighted Gene Coexpression Network Analysis (WGCNA) has proved to be a powerful tool, especially in constructing coexpression modules based on the relationship between genes as well as identifying hub genes. 14 –21 It has also been used recently to identify the biological processes, key hub genes, and biological functions of certain genes linked to cancers. 22 –24 Compared with other bioinformatic analysis methods, WGCNA is advantageous in that it can transform gene expression data into coexpression networks (modules); it can also provide insights into signaling networks. 25,26 With this transformation, a clearer view of the molecular mechanism behind the disease can be provided.
This study aimed to use a bioinformatics method to identify genes with novel and promising HCC prognosis biomarkers and possibly targeted therapies. They also analyzed the association of survival outcomes with the expression levels of these genes. Once these genes are validated to be effective prognosis biomarkers and targeted therapies, certain clinical interventions could be applied to improve HCC patients' outcomes.
Methods
Data acquisition and identification of differentially expressed genes
mRNA expression profiles were downloaded from Broad GDAC (where the data come from TCGA project), a publicly available human cancer database. Nine normal controls and 17 liver cancer samples were included in the analysis. The differentially expressed genes (DEGs) were identified using R software and DESeq2 R package. The thresholds were restricted as follows: fold change ≥2.0; p value ≤0.05; RPKM (Reads Per Kilobase per Million mapped reads) ≥2.5.
WGCNA to identify hub genes
WGCNA was performed to construct coexpression modules in normal samples and tumor samples. Module genes are groups of genes with similar patterns of expression. 27
The authors first constructed a weighted correlation network by creating a matrix of pairwise correlations between all DEGs. The function of soft connectivity from package WGCNA was used to evaluate the scale independence and the mean connectivity degree of different modules with different power value whose range was from 1 to 30. The lowest power value was determined for constructing network when the scale-free topology fit value was 0.8. Next, the linkage hierarchical clustering was produced by the topological overlap measure to carry out module detection and acquire the cluster dendrogram. The Dynamic Tree Cut algorithm was used to cut the cluster dendrogram and define the coexpression clusters of DEGs as modules. The interactions between modules were visualized as a heat map.
A hub gene is a gene with high connectivity inside a coexpression module. In the WGCNA analysis, genes with a kME value >0.9 were put in one module. kME is the module membership value of a module gene. They chose genes with kME >0.9 as members of a module. These genes can represent the expression pattern of the module.
GO and KEGG enrichment for total DEGs and module genes
The authors used enrichment R package to enrich the Gene ontology (GO) and KEGG terms for all DEGs. The Fisher's test was used to evaluate the significance of GO and KEGG enrichments. They then performed GO and KEGG enrichment for genes in the two most significant modules dependent on the Database for Annotation, Visualization, and Integrated Discovery (DAVID,
Construction of protein–protein interaction network for total DEGs and module genes
To construct the protein–protein interaction (PPI) network, the authors loaded the gene symbols of total DEGs or module genes to Metascape to visualize the PPI network. Cytoscape built-in of Metascape was used to produce the relationships between the enriched terms of total DEGs. The Molecular Complex Detection (MCODE) built-in was applied to identify densely connected network components and key genes of module genes.
Validation of significant hub genes' expression using GEO datasets
GSE101728 was downloaded from GEO Datasets (
Survival analysis
Kaplan–Meier Plotter (
Result
Identification, GO, and KEGG enrichment of total DEGs
The expression profiles of 10,054 genes in normal tissues (n = 9) and liver carcinoma tissues (n = 17) were selected to identify the DEGs and construct gene coexpression networks. Genes with RPKM (reads per kilobase per million mapped reads) ≥2.5 were selected. The screening of 1524 DEGs with fold changes >2 and p < 0.05 was then carried out using DESeq2 R package for the enrichment analysis and WGCNA. The top 20 upregulated genes and downregulated genes are shown in the heat map in Figure 1A. After the screening process, R software was used to analyze the GO and KEGG enrichment for all DEGs. Metabolic and catabolic processes were the key GO terms (Fig. 1B). The key KEGG pathways included the degradation and metabolism of substrates such as fatty acid, amino acids, carbohydrate etc., cell cycle, and DNA replication (Fig. 1C).

Identification and functional enrichment of total DEG.
The Cytoscape built-in of Metascape software was then used to enrich the terms and analyze the relationships between the enriched terms. Findings indicated that metabolic processes were included in the top enriched terms, such as monocarboxylic acid metabolic process, amino acids (specifically valine, leucine, and isoleucine) degradation, and steroid metabolic process (Fig. 1D).
Typically, metabolic processes include catabolism and anabolism processes, which constantly happen in both healthy and cancerous cells. These processes help meet the bioenergetic, biosynthetic, and redox demands of cancerous cells. The hallmark of cancer is a metabolism that is highly active, and these top enriched metabolic processes suggest that metabolism is very active in HCC. This suggestion means that HCC requires plenty of energy and nutrients. These results provide an overview of the fold change values, the enriched terms of total DEGs, and the relationships between the terms.
Identification of significant modules in tumor tissues
WGCNA was performed to identify the coexpression network of total DEGs. This unsupervised and unbiased analysis provided a weighted correlation network corresponding to closely correlated DEG clusters. Classified into one module were genes with similar expression patterns. Most of the total DEGs were clustered into colored (red, green, and blue) modules (Fig. 2A).
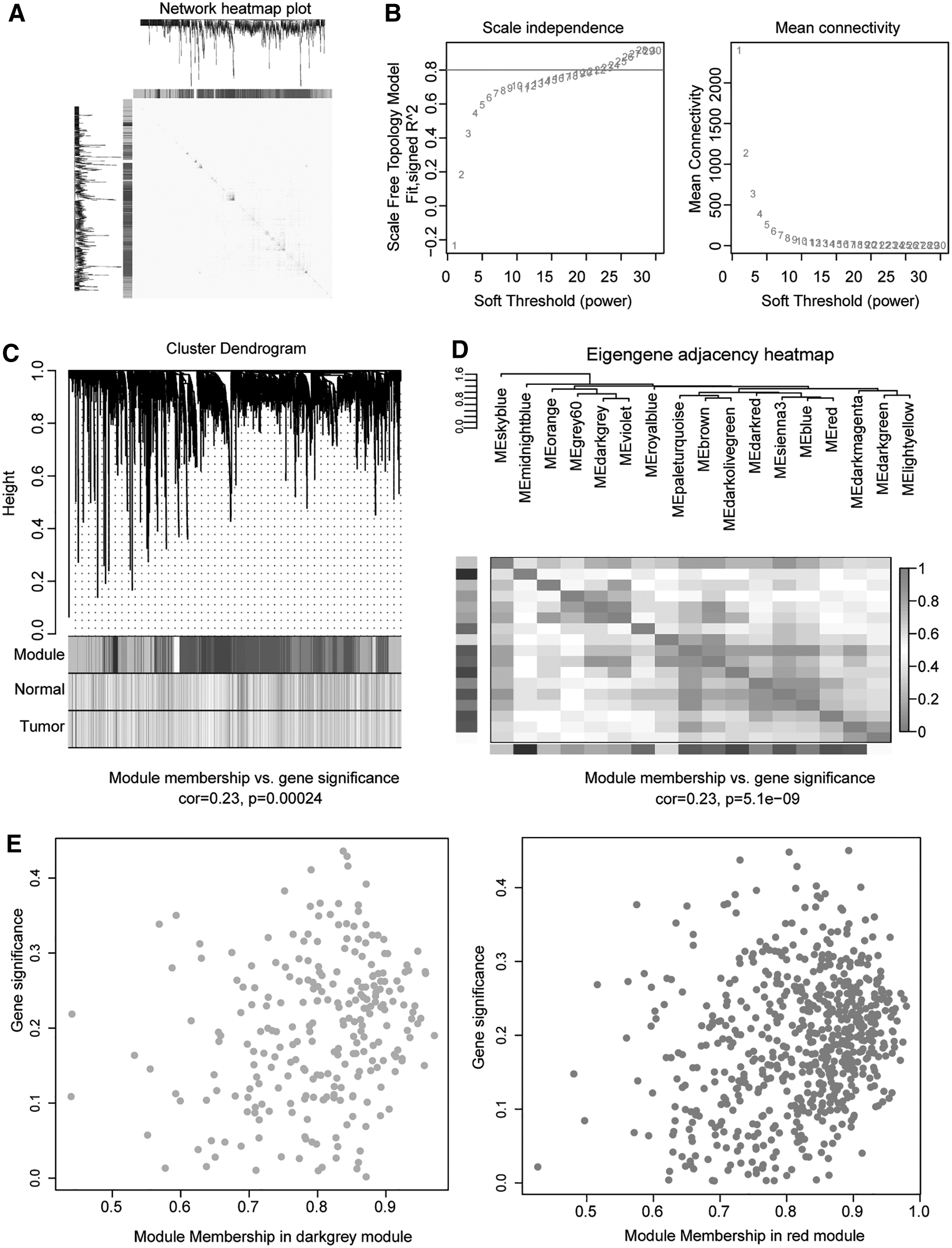
Identification of significant modules in HCC using WGCNA.
The network topology for various power values was analyzed to balance the scale independence and mean connectivity of WGCNA. The lowest power value, which was 22, was used to produce a hierarchical cluster dendrogram (Fig. 2B, C). The correlation between 17 significant modules was measured based on the calculated eigengene adjacency between the modules (Fig. 2D). The largest module of these 17 modules contained 1342 genes, while the smallest module included 59 genes. The dark gray and red modules were chosen for further GO and KEGG enrichment based on the number of genes and the correlation significance. The correlations of the module membership and gene significance are illustrated in Figure 2E.
GO and KEGG enrichment analysis of module genes
The authors loaded the genes with kME >0.9 in dark gray and red modules into the online DAVID database to analyze the GO and KEGG enrichments. The GO enrichment results showed that most of the genes in the dark gray module were located in mitochondrial inner membrane involved in mitochondrion-mediated processes such as mitochondrial translational elongation, mitochondrial translational termination, and mitochondrial electron transport NADH to ubiquinone, and so on. These processes are instrumental to molecular functions, including structural constituents of the ribosome, NADH dehydrogenase (ubiquinone) activities, and protein binding (Fig. 3A).
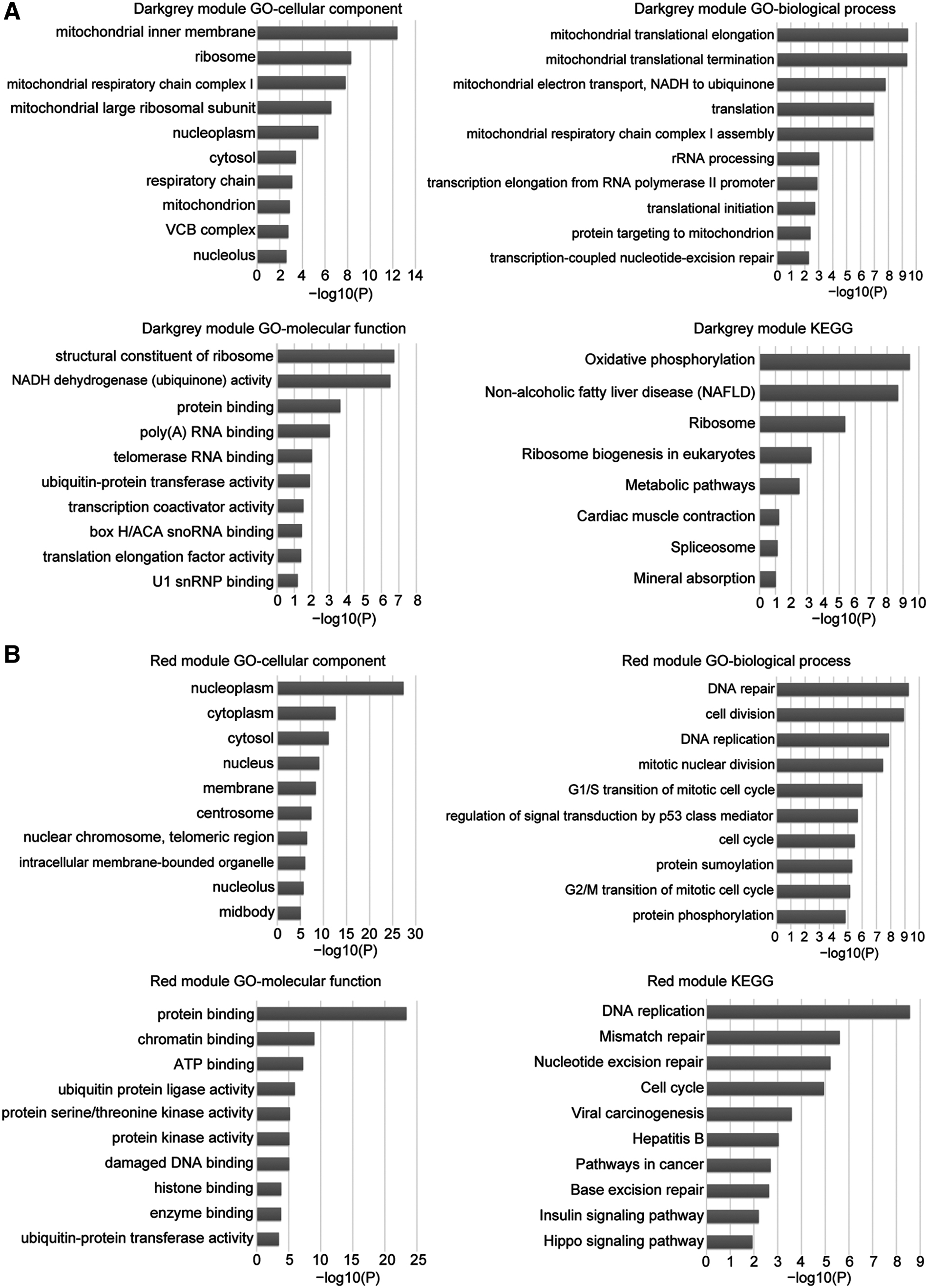
GO and KEGG enrichment analysis of the dark gray and red module genes.
In addition, KEGG pathway enrichment results showed that oxidative phosphorylation, ribosome, and metabolic pathways were the top enriched pathway in the dark gray module (Fig. 3A). Perhaps this is because mitochondrion mediates a plethora of metabolic and catabolic processes such as ATP synthesis and glucose break down. Besides, oxidative phosphorylation takes place in mitochondria during the ATP formation process. Mitochondrial ribosomes also participate in the translation of mitochondrial mRNAs.
The analysis results of dark gray module genes were consistent with the enrichment analyses of all DEGs. However, the red module genes were primarily located on the nucleoplasm to carry out cell division, DNA repair, and DNA replication biological processes probably by binding to proteins, chromatins, and ATP (Fig. 3B). The top enriched KEGG pathways involved DNA replication, mismatch repair, nucleotide excision repair, and the cell cycle (Fig. 3B). Therefore, they speculated that the genes in the dark gray module might affect the pathogenesis of liver carcinoma by regulating mitochondrion-mediated biological processes and signaling pathways. Genes in the red module, nonetheless, might affect the pathogenesis of liver carcinoma by regulating DNA replication, mismatch repair, and the cell cycle.
PPI network construction of dark gray and red module genes and identification of the significant genes in the two modules
The PPI network and MCODE clusters of the dark gray module genes are presented in Figure 4A. The dark gray and red module genes were loaded into Metascape for PPI network construction. The top GO enrichment shown to interact closely were the ATP synthesis coupled electron transport and mitochondrial translational activities, including mitochondrial translational initiation, elongation, and termination. This finding is consistent with DAVID analysis results. In addition, the PPI network and MCODE clusters of the red module genes are presented in Figure 4B. Significant processes were the cell division and mitosis. Red module genes engaged in these processes probably by participating in sister chromatid cohesion and separation, DNA repair, and DNA's binding to proteins during the mitotic prometaphase.
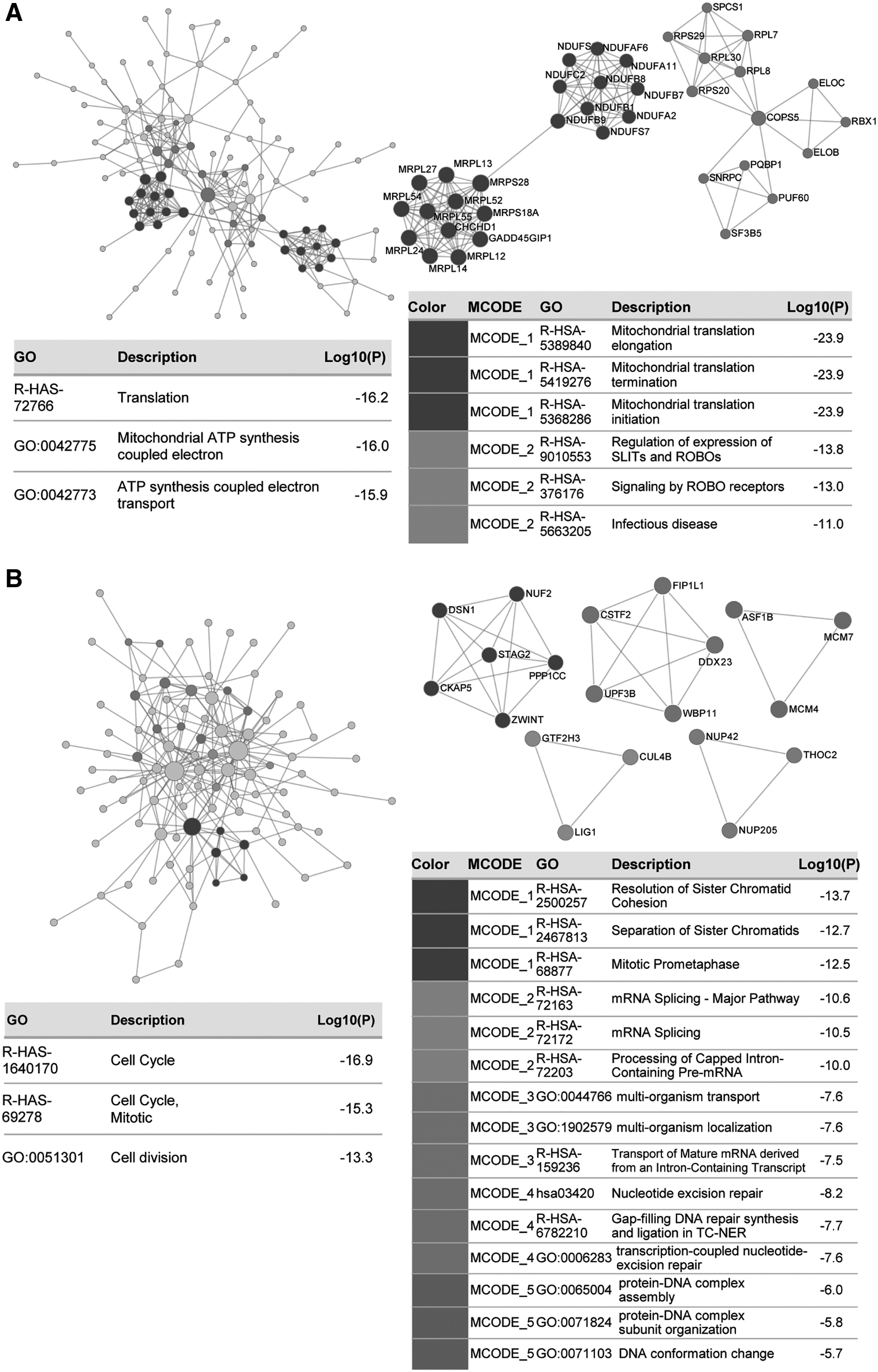
The construction of PPI network using Metascape.
Genes in the most significant MCODE clusters of the two module genes were chosen for the following prognosis analysis: NDUFC2, NDUFS7, NDUFB1, NDUFB9, NDUFA2, NDUFB7, NDUFA11, NDUFAF6, NDUFS6, NDUFB8, MRPS28, MRPS18A, MRPL14, MRPL12, MRPL54, MRPL55, MRPL52, MRPL13, MRPL27, MRPL24, GADD45GIP1, and CHCHD1 in dark gray module; NUF2, DSN1, STAG2, PPP1CC, CKAP5, and ZWINT in red module.
Prognosis analysis of MCODE cluster genes using the Kaplan–Meier method
The authors transferred the MCODE genes of dark gray and red modules to the online Kaplan–Meier survival analysis software. Results indicated unfavorable prognostic factors of overall survival for HCC patients, such as upregulated NDUFAF6 mRNA in the dark gray module and CKAP5, DSN1, NUF2, PPP1CC, and ZWINT mRNAs in the red module (Fig. 5). Their findings suggested that valuable prognosis biomarkers in HCC might include NDUFAF6, CKAP5, DSN1, NUF2, PPP1CC, and ZWINT.

The prognosis values of significant genes in dark gray and red modules. The survival outcome was analyzed using Kaplan–Meier Plotter online tool with 95% CIs of HR. Only those with significant difference were shown here. CI, confidence interval; HR, hazard ratio.
The expression of NDUFAF6, CKAP5, DSN1, NUF2, PPP1CC, and ZWINT in HCC
GSE101728 contains the expression profiling of mRNAs in seven HCC tumor tissues and seven liver adjacent tissues. It was used to validate the expression of such significant genes as NDUFAF6, CKAP5, DSN1, NUF2, PPP1CC, and ZWINT. The expression levels of NDUFAF6, CKAP5, DSN1, NUF2, and ZWINT in tumor tissues were significantly higher than in adjacent tissues, especially the expression level of ZWINT (P = 0.0003). Even though the expression of PPP1CC was upregulated in tumor tissues, no significant difference between the two groups was observed (p = 0.2089) (Fig. 6).

The validation of the expression of identified genes using GSE101728 datasets. GSE101728 included seven liver-adjacent tissues and seven liver tumor tissues. The expression levels of NDUFAF6, CKAP5, DSN1, NUF2, PPP1CC, and ZWINT in GSE101728 were measured, and the Student's t-test was used to analyze the significant differences between liver-adjacent and tumor tissues.
NUF2 and ZWINT have been proved as oncogenes in HCC. 29,30 Determined to use a novel model to identify potential HCC biomarkers and targeted therapies, the authors nominated NDUFAF6, CKAP5, and DSN1 as the key genes that required further investigation. Using their GO and KEGG enrichment analyses, they hypothesized that NDUFAF6 might participate in HCC in a mitochondrion-mediated manner possibly by regulating translational processes, whereas CKAP5 and DSN1 might take part in the cell cycle by regulating the cohesion and separation of sister chromatids during the prometaphase of mitosis. Because of the significant upregulation in HCC tumors, they might report them as potential oncogenic effectors that could be used as novel targeted therapies in HCC.
Discussion
Hundreds of thousands of people worldwide still die annually of HCC, which is one of the most common malignant cancers involving complex molecular regulations. 11 The aim of this article was to investigate all the valuable genes with the potential to become prognosis biomarkers and possible targeted therapies in HCC. The authors' observations reveal that NDUFAF6, CKAP5, and DSN1 might have this potential in HCC. Hence, the authors hypothesized that NDUFAF6 might participate in HCC by regulating mitochondrion-mediated translational processes, while CKAP5 and DSN1 might contribute to HCC by controlling the cohesion and separation of sister chromatids during the prometaphase of mitosis.
Regarded as a powerful extrachromosomal nuclear epigenetic system, a mitochondrion is an epigenetic origin of diseases, and it can retrograde control nuclear gene expression. 31,32 Otto Warburg was the first scholar to suggest that cancer is a mitochondrial metabolic disease. 33 This suggestion has been supported by growing pieces of evidence in the literature. 34,35 The loss of mitochondria or mitochondrial metabolism dysfunction impairs cell respiration, thereby leading to tumor cell phenotypes, including glycolysis, dedifferentiation, uncontrolled proliferation, aggressive mobility, and the formation of tumor microenvironments. 36,37 Mitochondrial dynamics of cancer cells is thereby affected by the tumor microenvironment's stress signals such as oxidative stress and energy deprivation, both of which are associated with cancer progression. 38
Research also reveals that the replacement of impaired mitochondria with normal mitochondria represses tumorigenicity, 39 probably by restoring normal metabolism in liver cancer. This revelation has been supported by the intrahepatic transplantation in adult syngeneic Fischer 344 rats, which led to the morphological differentiation of tumor cells. The authors claimed that hepatic cell-neoplastic hepatic cell fusion in the hepatic microenvironment suppressed tumorigenicity because cell fusion is a unique and physiological process in the rat's liver microenvironment. 40 What this means is that cell fusion might have introduced normal mitochondria into cancer cells, thus resulting in metabolic homeostasis.
Besides, mitochondrion-mediated processes are critical to all sorts of cell activities in HCC. This claim has been proved by previous studies. For instance, mitochondrial oxidative metabolism was reported to be closely correlated with liver carcinoma. 41,42 Lee et al. also noted that mitochondrial respiratory (oxidative phosphorylation) defects were an essential HCC bioenergetics feature. 37 In their study, Shen et al. revealed that the activation of mitochondrial oxidative phosphorylation played a key role in inducing the apoptosis in HCC. 43 As for the mitochondrial translational initiation, elongation, and termination, only a few studies have been conducted to investigate their relationship with HCC.
NDUFAF6 (NADH-Ubiquinone Oxidoreductase Complex Assembly Factor) is a critical gene that participates in mitochondrial translational processes. It encodes a protein that primarily localizes itself to the mitochondrion and nucleus and participates in the assembly of NADH-ubiquinone oxidoreductase of the mitochondrial respiratory chain by regulating the biogenesis of subunit ND1. Yet, this gene has never been studied in HCC. The authors thus hypothesized that NDUFAF6 might regulate HCC progression by regulating the mitochondria-mediated respiratory system, which provides energy for metabolism homeostasis and protein translation in mitochondria.
Also, mitochondria-mediated activities such as oxidative phosphorylation, DNA replication, DNA repair, and the cell cycle were also shown to be associated with cancer genesis. 44 –48 For instance, Puigvert et al. presented an overview of current and novel strategies to target DNA repair and replication to treat cancers. 49 Roy et al. also discussed the role of regulatory molecules related to the cell cycle in cancers such as cyclin-dependent kinase (CDK), cyclin D, and P53. 44 Keeping these studies in mind, they herein identified two novel cell cycle-related genes that could be novel prognosis biomarkers and targeted therapies in HCC, such as CKAP5 (Cytoskeleton Associated Protein 5) and DSN1.
CKAP5 encodes a cytoskeleton-associated protein, and it is widely expressed in the plasma membrane, cytoskeleton, nucleus, and cytosol. The N-terminal half of this protein contains a microtubule-binding domain, and the C-terminal half includes a KXGS motif that can bind tubulin dimers. This protein has two distinct roles in spindle formation. One, it is essential for centrosomal microtubule assembly; and two, it protects kinetochore microtubules from depolymerization.. 50,51 These roles may be necessary for the proper interaction of microtubules with the cell cortex for directional cell movement. In addition, DSN1 is a component of the minichromosomal instability-12 (MIS12) centromere complex, and it is required for proper kinetochore assembly and cell cycle progression.
Hypothetically, both CKAP5 and DSN1 are critical in regulating cell cycle progression. Yet, limited studies have explored how these two genes take part in cell cycle progression during HCC development. Nonetheless, CKAP5 was found to be involved in cancer development. For instance, by binding to ado-trastuzumab emtansine, CKAP5 caused cell membrane damage and led to calcium influx into the hepatocytes, thereby resulting in microtubule network disorder and apoptosis. 52 CKAP5 was also identified as an interacting protein of ARHGEF16, the knockdown of which led to a decrease in glioma cell migration and proliferation. 53 After several analyses, Schneider et al. found that CKAP5 was overexpressed in tumor tissues and associated with the prognosis of nonsmall cell lung cancer patients. 54
In terms of DSN1, it was reported to be upregulated in HCC tissues, and it was associated with the poor overall survival and disease-free survival outcomes of the patients. 55 Also reported in the literature is the relationship between DSN1 and the cell cycle in other human cancers. For instance, DSN1 was found to be significantly upregulated and associated with cell cycle progression. 56 The depletion of DSN1 resulted in G2/M phase arrest and impaired the migration, invasion, and anchorage-independent growth of colorectal adenoma cells. 57
Closely believed to be correlated with tumorigenesis is the cell cycle, which can be modulated by tumor microenvironments. In fact, a microenvironment plays an essential role in quiescence-proliferation regulation. 58,59 A cell, for instance, can instruct and modify its microenvironment by remodeling the extracellular matrix as well as physically and chemically cross-talking with its neighboring cells. 58 The viciously reciprocal influence between the cell and its microenvironment helps adjust all aspects of cell behaviors, including quiescence and proliferation. 58 Besides, the change of cellular microenvironment has been shown to influence cells to enter the cell cycle. 59 –61 A healthy microenvironment may prevent cells from penetrating the cell cycle, while a cancerous microenvironment may permit cells to reenter the cell cycle and proliferate uncontrollably.
In this study, CKAP5 and DSN1 were identified as critical players in cell cycle progression of HCC. Therefore, CKAP5 and DSN1 could be aberrantly regulated due to the microenvironment changes. They can be aberrant cell cycle progression markers. However, more research is needed to understand how HCC microenvironment regulates these two genes and what the roles of the two genes are.
Like many studies, this research is not immune to constraints. Certain limitations of the current analysis include a small size of the samples for the mRNA profiling. However, the results have been validated using GSE101728 datasets, a validation that makes the results more reliable and credible. In future work, a larger sample size for mRNA profiling or more validation datasets will be utilized. By validating the upregulated expression of these genes in HCC tissues, the authors speculated that these genes were like to be cancer drivers, not suppressers. This speculation needs confirmation, however. Hence, they recommend that in-depth studies should be conducted in future research to confirm the findings in the current study.
The follow-up of this current work will include the analysis of the association among their expression levels with HCC patients' characteristics, the effects of their knockdown on mitochondrion-mediated metabolisms, cell cycle progression, and in vivo tumorigenesis assays.
After verifying the effects of the three genes NDUFAF6, CKAP5, and DSN1 in HCC cell lines, cell lines-derived xenograft mice models will be used to study the effects of gene knockdown on tumor formation in vivo. More specifically, human HCC cells with NDUFAF6, CKAP5, and DSN1 knockdown will be injected subcutaneously to nude mice, and the tumor formation will be monitored to represent the effects of NDUFAF6, CKAP5, and DSN1 knockdown on tumor formation in vivo. Downstream analysis of the in vivo assays will also be done, including the tumor mass statistics, pathologic analysis (usually H&E staining) of the tumor, and the expression of the genes of interest (NDUFAF6, CKAP5, or DSN1) in tumor tissues.
In an effort to discover potential biomarkers and targeted therapies for HCC, the authors' found no drugs for NDUFAF6 and DSN1. However, they discovered two possible drugs for CKAP5. The two drugs are 10-DAB (10-Deacetylbaccatin) and ABT-751 (E7010). The former is an antineoplastic agent and an anticancer intermediate that targets cytoskeleton and the latter is a bioavailable tubulin-binding and antimitotic agent. However, no clinical trials have been registered, and no studies related to the two drugs and CKAP5 have been found. Nonetheless, they believe that a study on CKAP5 responding to the two drugs in cell experiments will be worthwhile.
More importantly, the authors identified three genes with the potential to be novel and promising HCC prognosis biomarkers and targeted therapies. The findings in the present study reveal a future direction for their in-depth studies in terms of the validation of potential HCC prognostic biomarkers and targeted therapies, including how these biomarkers regulate mitochondrion-mediated metabolism and cell cycle progression to affect HCC development. Thus, the authors believe that the current work sets a proper direction for them to carry out future experiments.
Conclusion
In summary, the authors report that NDUFAF6, CKAP5, and DSN1 may have the potential to be prognostic biomarkers and intervention targets in HCC because NDUFAF6 may participate in HCC pathogenesis by regulating mitochondrion-mediated translational processes, while CKAP5 and DSN1 may participate in HCC by controlling the cohesion and separation of sister chromatids during the prometaphase of mitosis.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.