
Abstract
Background:
Hepatocellular carcinoma (HCC) has high morbidity and mortality, but current therapeutic methods cannot effectively improve patient's prognosis. FOXD3-AS1, a new identified long noncoding RNA, is dysregulated in several cancers and functions as a carcinogenic or tumor-suppressor factor. However, the function of FOXD3-AS1 in HCC has not been reported.
Materials and Methods:
Quantitative real time-polymerase chain reaction was applied to evaluate the expression of FOXD3-AS1 in HCC tissues and cell lines. miRDB and TargetScan websites were utilized to predict the interaction network of FOXD3-AS1 as a competing endogenous RNA. The interaction was confirmed by luciferase reporter assay and RNA binding protein immunoprecipitation (RIP) assay. The effect of FOXD3-AS1 on HCC cells (Huh6) were measured by cell counting kit (CCK)-8, BrdU cell proliferation assay, Transwell invasion assay, and wound healing assay.
Results:
FOXD3-AS1 was overexpressed in HCC, and HCC patients with the high level of FOXD3-AS1 had a poor prognosis. In addition, FOXD3-AS1 knockdown considerably inhibited the proliferation, migration, and invasion of Huh6 cells. Besides, FOXD3-AS1 functioned as a sponge of miR-335, and RICTOR was a direct target gene of miR-335. Furthermore, FOXD3-AS1 could enhance the level of RICTOR through sponging miR-335. Moreover, the knockdown of FOXD3-AS1 could competitively bind with miR-335 to suppress RICTOR expression, thereby inhibiting the growth of Huh6 cells through the deactivation of AKT signaling pathway.
Conclusions:
FOXD3-AS1 is crucial for the tumorigenesis and progression of HCC. The interaction among FOXD3-AS1, miR-335, and RICTOR provides a novel insight for understanding the molecular mechanism of HCC, and FOXD3-AS1, miR-335, and RICTOR can be regarded as the potential targets for HCC treatment.
Introduction
Hepatocellular carcinoma (HCC) accounts for >80% of primary liver cancers and is currently estimated to be the fourth most frequent cause of cancer-related death worldwide. 1,2 HCC represents a major public health problem, caused by metabolic liver disease, alcohol abuse, hepatitis C virus infection, and hepatitis B virus infection. 3,4 Surveillance is used to diagnose HCC patients at an early stage, and those patients can be curatively treated by liver transplantation, surgical resection, or local ablation. 5,6 Nevertheless, most patients with HCC are diagnosed at an advanced stage, and sorafenib is regarded as the only effective treatment in advanced HCC. 7 Nevertheless, treatment with sorafenib increases the median of overall survival times only from 8 to 11 months. 8 Thus, novel therapeutic methods for HCC are still needed.
The carcinogenesis of HCC is a very complex process that is mediated by gene mutations, chromosomal aberrations, diverse genetic and epigenetic alterations, and aberrant molecular pathways. 9 The molecular mechanism underlying HCC has not been well understood, and thus further exploring its molecular mechanism can contribute to developing therapeutic methods of HCC. 10 Long noncoding RNAs (lncRNAs, >200 nucleotides) do not encode proteins, but have been found to be key regulators in various biological and cellular processes affecting cell cycle progression, proliferation, migration, and survival. 11 –13 Increasing evidence suggests that lncRNAs are aberrantly expressed in diverse types of cancers, and are crucial for the carcinogenesis of the cancer. 14,15 FOXD3-AS1 (lncRNA forkhead box D3 antisense RNA 1), a novel identified lncRNA, is the antisense transcript of FOXD3. 16 FOXD3-AS1 has been reported to be a tumor promoter in glioma 16 and cutaneous malignant melanoma, 17 and yet exerts tumor-suppressive roles in neuroblastoma. 18 However, the function of FOXD3-AS1 in HCC has not been reported.
Therefore, the work studied the expression level and functional roles of FOXD3-AS1 in HCC. Based on the competing endogenous RNA (ceRNA) hypothesis, the study identified miR-335 binding to FOXD3-AS1 and observed RICTOR is a target of miR-335. The authors further studied the interaction among FOXD3-AS1, miR-335, and RICTOR, and explored the underlying mechanism of FOXD3-AS1 in HCC cells.
Materials and Methods
Patient tissue specimens
The selected HCC patients (a total of 68) underwent curative surgery at Affiliated Hospital of Hebei University, and they did not receive radiotherapy or chemotherapy before resection. Written informed consent was obtained from all participants. In this study, the use of human specimens was approved by the Ethics Committee of Affiliated Hospital of Hebei University.
Cell culture and transfection
Human normal liver cells (HL-7702) and HCC cell lines (Huh7, Huh6, SK-HEP-1) were purchased from Cell Bank of Chinese Academy of Sciences, and were incubated in RPMI 1640 medium (Hyclone) containing 10% fetal bovine serum (FBS), 50 μg/mL streptomycin (Gibco), and 100 mg/mL penicillin (Gibco) under a humidified atmosphere at 37°C with 5% CO2.
For transfection, Huh6 cells were transfected with 100 nM FOXD3-AS1 shRNAs (FOXD3-AS1 sh1#, FOXD3-AS1 sh2#), scramble shRNAs (control group), miR-335 mimic, miRNA negative control (miR-NC), miR-335 inhibitor, and inhibitor negative control (NC inh) using Lipofectamine 3000 according to the manufacturer's protocol. FOXD3-AS1 shRNAs and scramble shRNAs were synthesized by GenePharma Co., Ltd. (Shanghai, China), and the other vectors were purchased from Invitrogen (Thermo Fisher Scientific).
Quantitative real time-polymerase chain reaction
Total RNA from HCC cell lines (5 × 106 cells/mL) and tissues (50 mg) was extracted with TRIzol Reagent (1 mL; Thermo Fisher Scientific). After the quantification of RNA concentration using spectrophotometry on the NanoDrop 2000 (Thermo Fisher Scientific), reverse transcription of total RNA (1 μg) was performed using Prime Script RT Master Mix (Takara, Japan) to obtain cDNA.
For miR-335 detection, miRNA was extracted using miRNeasy Mini kit (Qiagen GmbH), and cDNA synthesis was conducted through the TaqMan microRNA Reverse Transcription Kit (Thermo Fisher Scientific) according to the manufacturer's protocol.
cDNA was used as template, and the primers were obtained from Bioengineering (Shanghai, China). The primer sequences are listed in Table 1. Quantitative real time-polymerase chain reaction (qRT-PCR) was performed on an ABI 7500 Fast Real-Time PCR system using a SYBR® Premix Ex Taq™ RT-PCR Kit (Takara Biotechnology Co., Ltd.). The amplification reaction conditions are as follows: 95°C for 5 min (precycling), 40 cycles of 95°C for 30 s (denaturation), 60°C for 30 s (annealing), 72°C for 30 s (extension), and 75°C for 1 s (postextension). Relative expression level was evaluated using the 2−ΔΔCq method and normalized to two reference genes (β-actin and U6).
The Sequences of the Primers
Cell counting kit-8
Twenty-four hours after transfection, the cells were plated into 96-well plates (cell density, 1 × 104 cells per well) and then cultured in RPMI 1640 medium with 10% FBS for indicated time points (24, 48, 72, and 96 h), followed by adding cell counting kit (CCK)-8 solution (10 μL; Engreen Biosystem Co, Ltd., Beijing, China). Next, the incubation was continued at 37°C for a further 4 h. Optical density was measured through a microplate reader at 450 nm.
BrdU cell proliferation assay
The transfected cells were seeded in 24-well plates (cell number, 1 × 104 cells per well) and incubated for 48 h in RPMI 1640 medium containing 10% FBS. After removing RPMI 1640 medium from cells, BrdU labeling solution (Thermo Fisher Scientific) was added to the plate well. Next, the cells were incubated at 37°C for 2 h, removed the labeling solution, and washed with phosphate buffered saline (PBS). Subsequently, the cells were fixed with 3.7% formaldehyde in PBS for 15 min at room temperature and treated with 0.2% Triton X-100 permeabilization buffer at room temperature for 10 min. After washing with PBS, the cells were added with anti-BrdU primary antibody and cultured at 4°C overnight. After removing solution and washing with PBS, DAPI was added into the plates and incubated with the cells at room temperature for 30 min. Finally, the cells were imaged by a light microscope.
Wound healing assay
After 24 h of transection, the cells were seeded in six-well plates followed by incubating in RPMI 1640 medium with FBS to 100% confluence. Then, the scratch was made through a sterile pipette tip (100 μL) through the cell monolayer. The debris was removed and the cells were incubated for another 24 h in serum-free RPMI 1640 medium at 37°C under 5% CO2. The cells were visualized by an Olympus BX51 light microscope (magnification, 100 × ; Olympus Corporation). The quantification of wound closure was performed by ImageJ software (v1.8.0).
Transwell invasion assay
After 24 h of transfection, cells were collected, suspended in serum-free RPMI 1640 medium, and then added to the upper chamber of Transwell coated with Matrigel matrix (Coring). The RPMI 1640 medium with 10% FBS was added to the lower Transwell chamber. After the cells were incubated at 37°C for 24 h, the invaded cells were fixed with 4% paraformaldehyde for 30 min at room temperature, followed by staining with 0.1% crystal violet at room temperature for 20 min. After washing with PBS, the samples were photographed under a light microscope (Olympus BX51; magnification, 200 × ).
Luciferase reporter assay
miRDB was utilized to identify the potential target miRNA of FOXD3-AS1, and TargetScan was applied to analyze the predicted target gene of miR-335. The 3′-untranslated region (UTR) fragment of FOXD3-AS1 or RICTOR wild type (WT) with the putative sites of miR-335 was amplified using PCR and then cloned into pGL3 vector (Promega, Madison), thereby obtaining FOXD3-AS1-WT or RICTOR-WT luciferase reporter vector. The corresponding 3′-UTR of FOXD3-AS1 or RICTOR mutation was also performed the same experimental procedure to obtain FOXD3-AS1-MUT or RICTOR-MUT control vector. These vectors with miR-335 mimic or miRNA control were, respectively, transfected into HCC cells using Lipofectamine 3000. After 48 h of incubation at 37°C, the luciferase activities of the samples were determined by a Dual Luciferase Reporter Assay kit (Promega) and normalized to the luciferase activity of Renilla.
RNA binding protein immunoprecipitation assay
RNA binding protein immunoprecipitation (RIP) assay was utilized to explore the interaction of FOXD3-AS1 with miR-335. Huh6 cells were lysed and then cultured with the conjugated anti-human Ago2 antibodies or anti-mouse IgG-magnetic beads in RIP buffer. The precipitated RNA was isolated and quantified by qRT-PCR to obtain the enrichment value.
Western blot
RICTOR (Cat. No. #2114; 1:1000 dilution), p-AKT (Cat. No. #4060; 1:2000 dilution), AKT (Cat. No. #4691; 1:1000 dilution), PCNA (Cat. No. #13110; 1:1000 dilution), E-cadherin (Cat. No. #3195; 1:1000 dilution), and GAPDH (Cat. No. #5174; 1:1000 dilution) were purchased from Cell Signaling Technology. Horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibodies (Cat. No. SA00001-2, 1:2000 dilution) were obtained from ProteinTech Group, Inc. Protein isolates were obtained from the lysates of Huh6 cells using RIPA lysis buffer (Beyotime, Shanghai, China), quantified by Bicinchoninic Acid Assay, and then separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Next, the protein was transferred to the polyvinylidene difluoride membranes, which were blocked with 5% skimmed milk at room temperature for 1 h and incubated with primary antibodies against RICTOR, p-AKT, AKT, PCNA, E-cadherin, and GAPDH overnight at 4°C. After washing by TBST buffer containing 0.05% Tween-20, the membranes were incubated with HRP-conjugated secondary antibodies for a further 1 h at room temperature. After rinsing with blocking solution, the membranes were visualized by ECL (Immun-Star™ HRP chemiluminescent detection kit; Bio-Rad Laboratories, Inc.) and the semiquantitative of the protein expression level was evaluated by ImageJ software (v1.8.0). GAPDH was applied as a loading control.
Statistical analysis
The experiments in this study were performed three independent times. All data were analyzed by GraphPad Prism 7 software and expressed as mean ± standard deviation. Statistical analysis between two groups was conducted by Student's t-test. The comparison among multiple groups was evaluated using one-way analysis of variance (ANOVA) followed by Tukey's test. Overall survival analysis was assessed through Kaplan–Meier method with log-rank test. The difference among groups was regarded as statistically significant when p-value <0.05.
Results
The overexpression of FOXD3-AS1 in HCC tissues and cell lines
The expression level of FOXD3-AS1 was determined by qRT-PCR. As compared with the adjacent normal tissues, the aberrant high expression of FOXD3-AS1 was observed in HCC tissues (Fig. 1A, p < 0.0001). In addition, the overexpression of FOXD3-AS1 was confirmed in HCC cell lines (Huh7, Huh6, and SK-HEP-1) when compared with normal liver cells (HL-7702) (Fig. 1C; SK-HEP-1 vs. HL-7702, p = 0.0011; Huh7 vs. HL-7702, p = 0.0002; Huh6 vs. HL-7702, p < 0.0001). Besides, Huh6 cells were selected for subsequent experiments because of the highest expression value of FOXD3-AS1 in Huh6 as compared with other HCC cell lines.
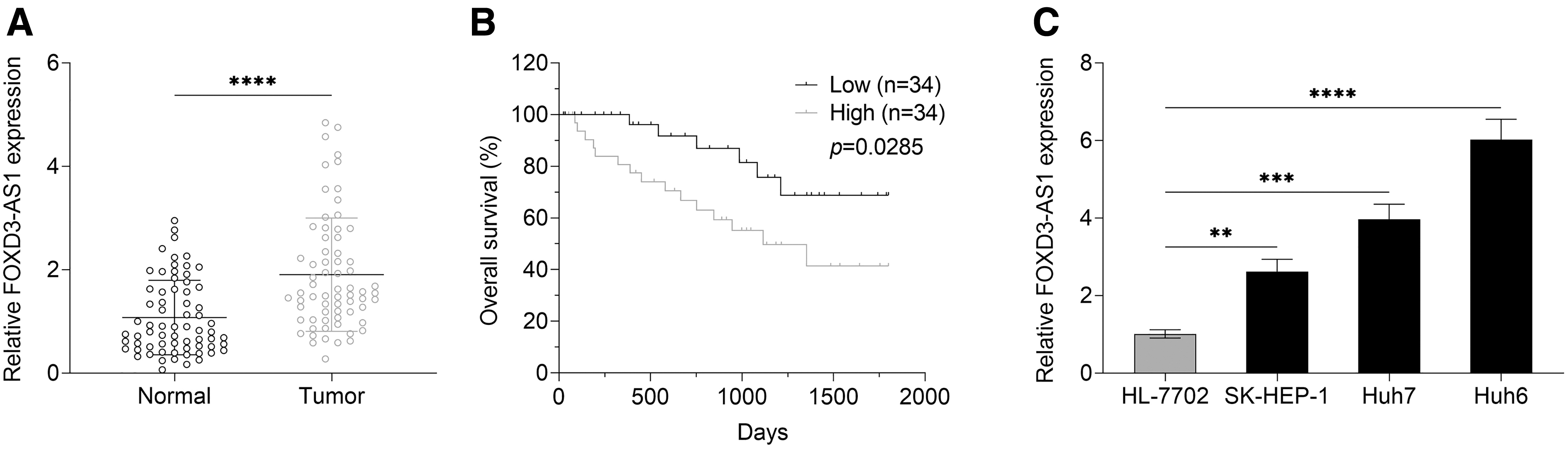
The overexpression of FOXD3-AS1 in HCC tissues and cell lines.
Moreover, FOXD3-AS1's median expression value was utilized to divide HCC patients into the FOXD3-AS1 low expression group and FOXD3-AS1 high expression group. Overall survival analysis in Figure 1B suggested that HCC patients with the high level of FOXD3-AS1 had a poor prognosis as compared with HCC patients in the FOXD3-AS1 low expression group.
The inhibition of the proliferation, migration, and invasion of HCC cells induced by FOXD3-AS1 knockdown
To explore the roles of FOXD3-AS1 in HCC cells, the proliferation, migration, and invasion of HCC cells were determined after FOXD3-AS1 knockdown. First, FOXD3-AS1 knockdown significantly downregulated the level of FOXD3-AS1 in Huh6 cells as compared with scramble control group and, therefore, the transfection of FOXD3-AS1 was effective (Fig. 2A; sh1# vs. Scramble, p = 0.0002; sh2# vs. Scramble, p < 0.0001). For cell proliferation detection, CCK-8 and BrdU cell proliferation assay were performed in this study. Two types of cell proliferation assays showed that the transfection of FOXD3-AS1 shRNAs (FOXD3-AS1 sh1#, FOXD3-AS1 sh2#) obviously inhibited the proliferation of Huh6 cells in comparison with the scramble group (Fig. 2B, two-way ANOVA, p < 0.0001; Fig. 2C, sh1# vs. Scramble, p = 0.0011; sh2# vs. Scramble, p = 0.0004). For cell migration and invasion detection, wound healing assay proved that FOXD3-AS1 knockdown effectively suppressed the migration of Huh6 cells (Fig. 2D, sh1# vs. Scramble, p = 0.0199; sh2# vs. Scramble, p = 0.0014), and Transwell invasion assay suggested that the invasion of Huh6 cells was inhibited by FOXD3-AS1 knockdown (Fig. 2E, sh1# vs. Scramble, p = 0.0012; sh2# vs. Scramble, p = 0.0001).
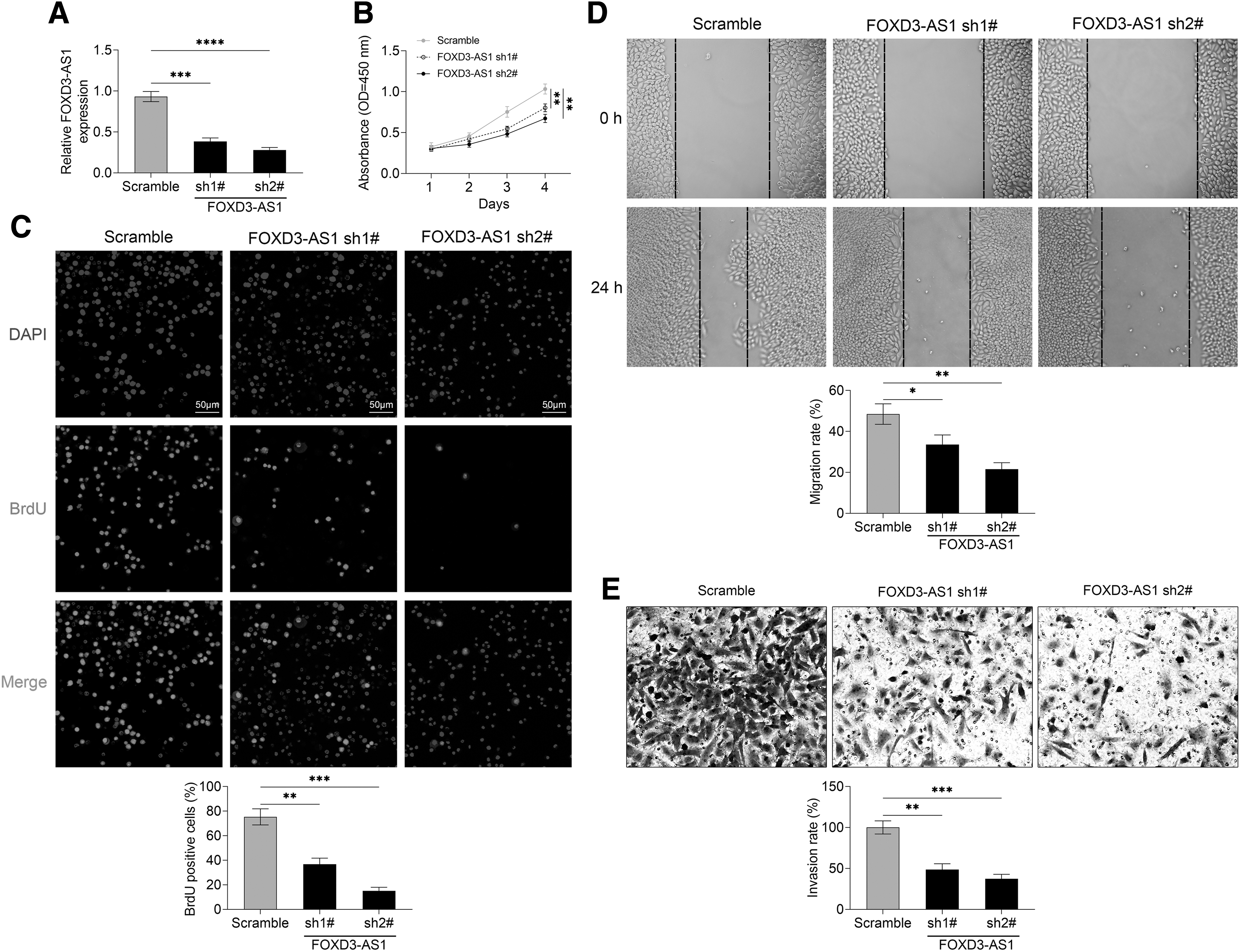
FOXD3-AS1 knockdown inhibits the proliferation, migration, and invasion of HCC cells.
FOXD3-AS1 functions as a sponge of miR-335 in HCC cells
Inspired by “competing endogenous RNA (ceRNA) hypothesis,” miRDB website (
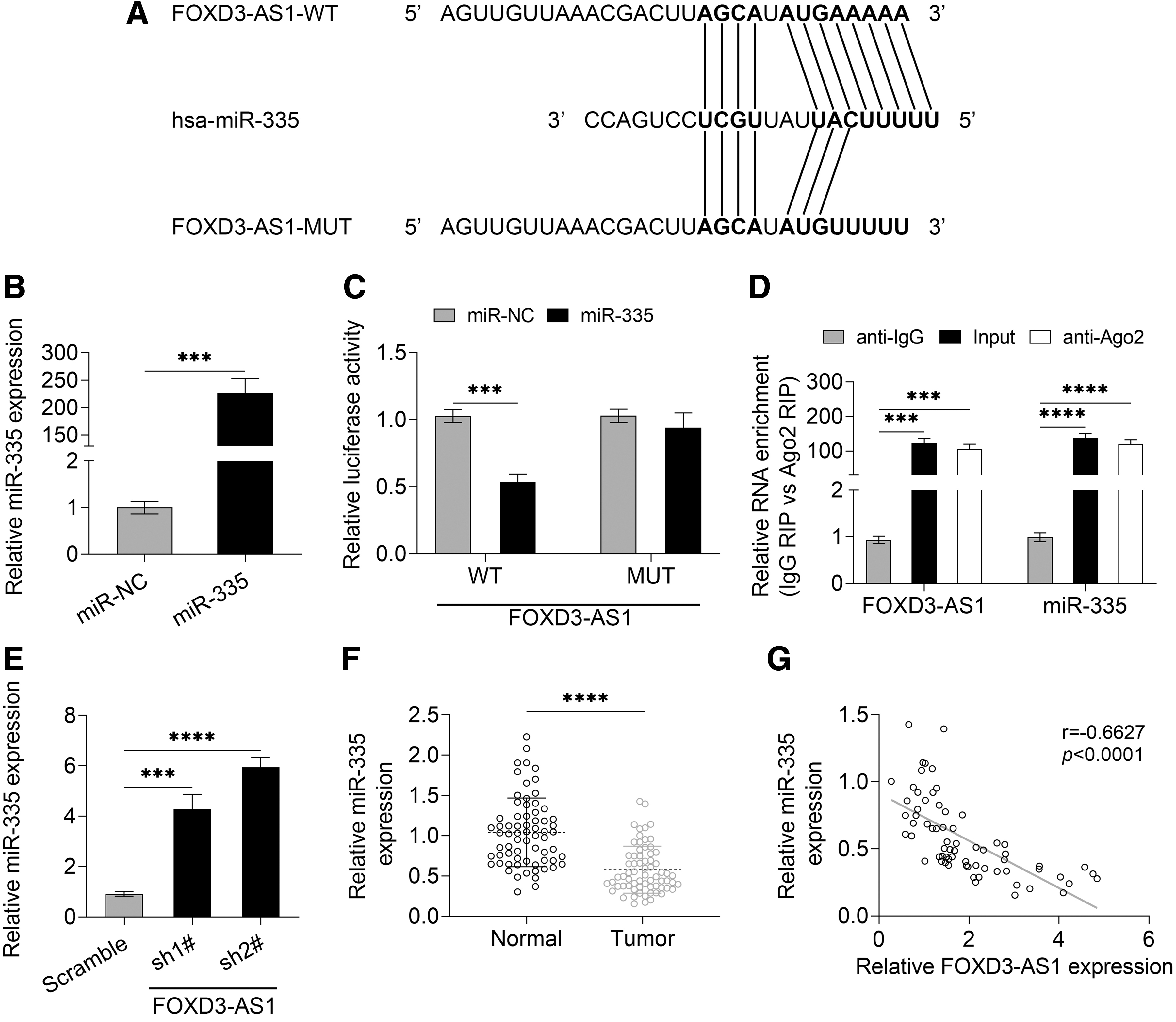
FOXD3-AS1 functions as a sponge of miR-335 in HCC cells.
Luciferase reporter assay and RIP assay indicated that FOXD3-AS1 directly targeted miR-335 in Huh6 cells (Fig. 3C, WT group, p = 0.000321; Fig. 3D, all p < 0.001 vs. anti-lgG). Moreover, Huh6 cells transfected with sh-FOXD3-AS1 showed a higher level of miR-335 in comparison with the control group (Fig. 3E, sh1# vs. Scramble, p = 0.0005; sh2# vs. Scramble, p < 0.0001). Moreover, the low expression level of miR-335 was observed in HCC tissues as compared with the normal tissues (Fig. 3F, p < 0.0001), and Pearson's linear regression analysis indicated a negative correlation between FOXD3-AS1 and miR-335 (Fig. 3G). Thus, FOXD3-AS1 could negatively regulate miR-335 expression and function as a sponge of miR-335.
FOXD3-AS1 upregulates the level of RICTOR through sponging miR-335
To further investigate the molecular mechanism of FOXD3-AS1, TargetScan website (
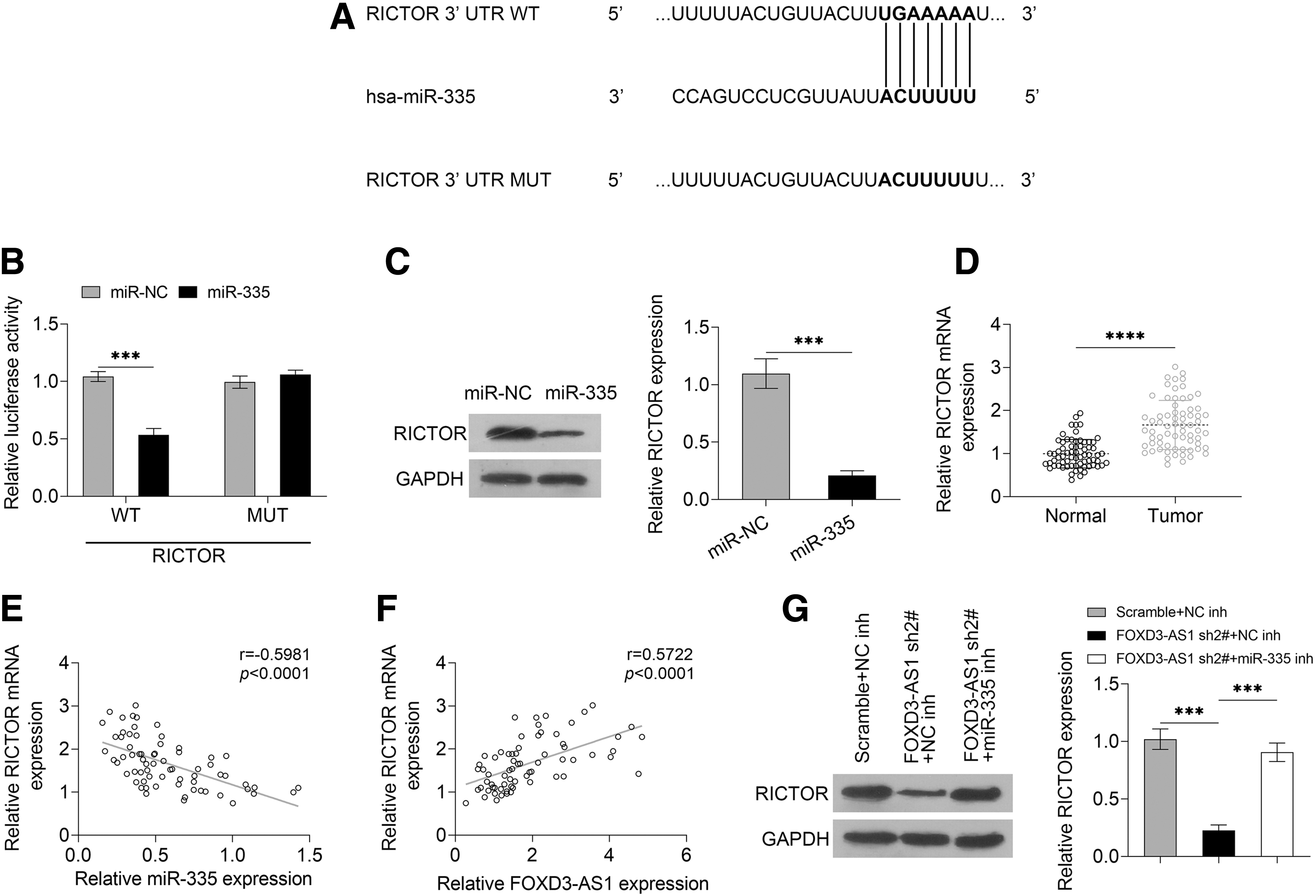
FOXD3-AS1 upregulates the level of RICTOR through sponging miR-335.
Moreover, the level of RICTOR showed a high level in HCC tissues as compared with the normal tissues (Fig. 4D, p < 0.0001). RICTOR had a positive correlation with FOXD3-AS1 and a negative correlation with miR-335, which was proved by Pearson's linear regression analysis (Fig. 4E, F). The knockdown of FOXD3-AS greatly downregulated the level of RICTOR, and the situation was reversed by the introduction of miR-335 inhibitor (Fig. 4G, FOXD3-AS1 sh2#+NC inh vs. Scramble+NC inh, p = 0.0002; FOXD3-AS1 sh2#+miR-335 inh vs. FOXD3-AS1 sh2#+NC inh, p = 0.0002). Therefore, the results speculated that FOXD3-AS1 upregulated the level of RICTOR through sponging miR-335.
FOXD3-AS1 mediates RICTOR expression to affect the proliferation, migration, and invasion of HCC cells through regulating the AKT signaling pathway
Based on the earlier findings, the authors speculated that FOXD3-AS1 might sponge miR-335 to regulate RICTOR expression, thereby affecting the proliferation, migration, and invasion of HCC cells. To test this inference, the authors performed CCK-8, BrdU cell proliferation assay, Transwell invasion assay, and wound healing assay. The results of CCK-8 and BrdU cell proliferation assay suggested that the inhibition of Huh6 cell proliferation induced by FOXD3-AS1 knockdown was reversed by the introduction of RICTOR (Fig. 5A, two-way ANOVA, p < 0.0001; Fig. 5B, FOXD3-AS1 sh2#+Vector vs. Scramble+Vector, p = 0.0002, FOXD3-AS1 sh2#+RICTOR vs. FOXD3-AS1 sh2#+Vector, p = 0.0003). The migration and invasion ability of Huh6 cell was suppressed by FOXD3-AS1 knockdown, which was changed by the co-transfection of RICTOR with FOXD3-AS1 shRNA (Fig. 5C, FOXD3-AS1 sh2#+Vector vs. Scramble+Vector, p = 0.0015, FOXD3-AS1 sh2#+RICTOR vs. FOXD3-AS1 sh2#+Vector, p = 0.0011; Fig. 5D, FOXD3-AS1 sh2#+Vector vs. Scramble+Vector, p = 0.0001, FOXD3-AS1 sh2#+RICTOR vs. FOXD3-AS1 sh2#+Vector, p = 0.0012).
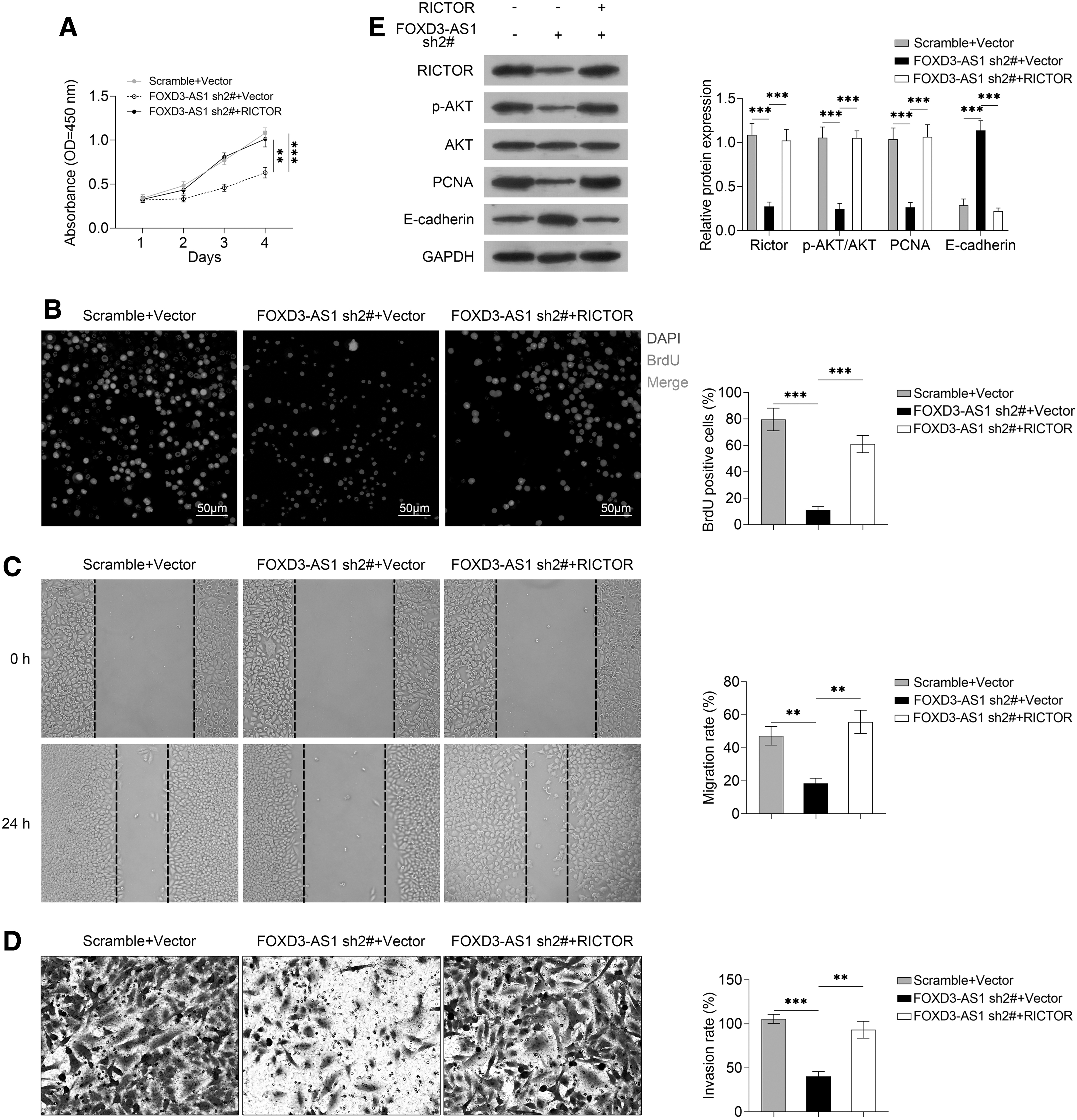
FOXD3-AS1 mediates RICTOR expression to affect the proliferation, migration, and invasion of HCC cells through regulating the AKT signaling pathway. After Huh6 cells were transfected with FOXD3-AS1 sh2#+RICTOR vector, FOXD3-AS1 sh2#+control vector, scramble control shRNA+control vector, the proliferation of the cells were measured by
On the one hand, RICTOR is known as a key regulator of the AKT pathway that is critical for the proliferation, migration, and invasion of cells. On the other hand, FOXD3-AS1 could competitively bind with miR-335 to upregulate RICTOR expression. Hence, the authors determined the changes of the AKT pathway to further explore the molecular mechanism of FOXD3-AS1 in HCC cells. As shown in Figure 5E, the decrease of p-AKT and PCNA and the increase of E-cadherin induced by FOXD3-AS1 knockdown were reversed by the introduction of RICTOR (all p < 0.001). Therefore, the knockdown of FOXD3-AS1 could inhibit RICTOR expression by competitively binding with miR-335, and thus deactivate the AKT signaling pathway to suppress the proliferation, migration, and invasion of Huh6 cells.
Discussion
The study indicated the overexpression of FOXD3-AS1 in HCC tissues and cell lines, and HCC patients with the high expression of FOXD3-AS1 showed a poor prognosis. In addition, FOXD3-AS1 knockdown considerably inhibited the proliferation, migration, and invasion of Huh6 cells. Besides, FOXD3-AS1 functioned as a sponge of miR-335, and RICTOR was a direct target gene of miR-335. RICTOR had a positive correlation with FOXD3-AS1 and a negative correlation with miR-335. Furthermore, FOXD3-AS1 could upregulate the level of RICTOR through sponging miR-335. Moreover, the knockdown of FOXD3-AS1 could suppress RICTOR expression, thereby inhibiting the growth of Huh6 cells through the deactivation of the AKT signaling pathway.
HCC has high morbidity and mortality, but current therapeutic methods cannot effectively improve patient's prognosis. 7,8 LncRNAs provide a novel insight for understanding the molecular mechanism of HCC, which can contribute to developing therapeutic methods of HCC. 10 FOXD3-AS1, a new identified lncRNA, has been found to be overexpressed in glioma and cutaneous malignant melanoma, and its high expression can promote the growth of the tumor cells. 16,17 Nevertheless, FOXD3-AS1 has also been reported to be downregulated in neuroblastoma, and its low expression promotes the progression of neuroblastoma. 18 As there is little research literature of FOXD3-AS1, the function of FOXD3-AS1 in various cancers will need to be explored. Especially, the roles of FOXD3-AS1 in HCC have not been reported. To explore this issue, the experiments in the study observed that FOXD3-AS1 was overexpressed in HCC tissues and cell lines. The high expression of FOXD3-AS1 was positively correlated with poor prognosis. Furthermore, FOXD3-AS1 knockdown effectively suppressed the proliferation, migration, and invasion of HCC Huh6 cells. Thus, FOXD3-AS1 could act as a tumor promoter in HCC.
The ceRNA hypothesis suggests that lncRNA can regulate gene expression through competing for shared miRNAs. 19 Based on the ceRNA hypothesis, the authors further studied the molecular mechanism of FOXD3-AS1 in HCC, and observed that FOXD3-AS1 functioned as a sponge of miR-335. Increasing evidence suggests that miR-335 is dysregulated in diverse cancers and is critical for the progression of tumors. 20 –22 Besides, miR-335 has been considered as a suppressor of different kinds of cancers such as gastric cancer and osteosarcoma. 23,24 In agreement with previous reports, the study found miR-335 had an aberrant low expression in HCC tissues as compared with the adjacent normal tissues. In addition, FOXD3-AS1 directly interacted with miR-335, and negatively regulated the expression of miR-335. Therefore, FOXD3-AS1 could function as a sponge of miR-335.
As miRNAs regulate gene expression to mediate various cellular processes, 25 the study identified RICTOR as a target gene of miR-335. RICTOR, the key component of the mTORC2 (mTOR complex 2), is required for the function of mTORC2. 26 RICTOR is upregulated in many cancers, which significantly induces the activation of AKT signaling pathway. 27,28 RICTOR as a key regulator of the AKT pathway plays critical roles in tumorigenesis and drives various essential processes such as cell migration, survival, differentiation, and proliferation. 29,30 This study also found the aberrant high expression of RICTOR in HCC. Moreover, the authors observed that the knockdown of FOXD3-AS1 could suppress the AKT signaling pathway activity to inhibit the proliferation, migration, and invasion of Huh6 cells through downregulating RICTOR expression.
Therefore, FOXD3-AS1 plays key roles in the tumorigenesis and progression of HCC. In addition, FOXD3-AS1 mediates the miR-335/RICTOR axis to affect the proliferation, migration, and invasion of HCC cells through regulating the activity of AKT signaling pathway, which offers a novel insight for understanding the molecular mechanism of HCC. FOXD3-AS1, miR-335, and RICTOR can be regarded as the potential therapeutic targets.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
Footnotes
Authors' Contributions
C.L. and R.Z. conceived and designed the experiments. M.Z. and J.S.Z. analyzed and interpreted the results of the experiments. X.S.Z., L.Z., M.D.Y., and X.X.Z. performed the experiments.
Ethics Approval and Consent to Participate
In this study, the use of human specimens was approved by the Ethics Committee of Affiliated Hospital of Hebei University. Written informed consent was obtained from all participants.
Disclosure Statement
There are no existing financial conflicts.
Funding Information
No funding was received for this article.