
Abstract
Background:
Gastric cancer (GC) is a common tumor found worldwide, and cisplatin is the first-line agent for the treatment of GC. However, the resistance to cisplatin is an obstacle. Here, we explored the biological mechanism of long noncoding RNA regulator of reprogramming (ROR) in the cisplatin resistance of GC.
Materials and Methods:
ROR, miR-519d-3p, and high mobility group protein A2 (HMGA2) expression in GC tissues and cells were measured by quantitative real-time polymerase chain reaction and Western blot. Cell viability, migration, invasion, and apoptosis were detected by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay, transwell assay, and flow cytometry, respectively. The relative protein expression was detected by Western blot. The interactions between miR-519d-3p and ROR, HMGA2 were predicted using miRcode and starBase v2.0 online database, and then verified by dual luciferase reporter assay and RNA immunoprecipitation assay. In addition, the xenograft tumor mouse model was constructed to verify the biological role of ROR in vivo.
Results:
The levels of ROR, HMGA2 were significantly upregulated, and miR-519d-3p was apparently downregulated in GC tissues and cells. The miRcode and starBase v2.0 online websites and dual luciferase reporter assay validated that miR-519d-3p directly interacted with ROR and HMGA2. Furthermore, ROR knockdown downregulated HMGA2 to restrain cell proliferation, migration, invasion, epithelial–mesenchymal transition (EMT), and cisplatin resistance in GC cells by targeting miR-519d-3p. In addition, the depletion of ROR repressed the xenograft tumor growth in vivo.
Conclusion:
In conclusion, we first found the ROR/miR-519d-3p/HMGA2 regulatory network to regulate cell proliferation, migration, invasion, EMT, and cisplatin resistance in GC, and this may shed light on the GC tumorigenesis.
Introduction
Gastric cancer (GC) is the most common malignant tumor over the world, especially in some East Asia countries including China. 1,2 Although some measures in detection and therapeutics have been improved, the survival time for GC patients is still short. 3 Cisplatin, an effective chemotherapy agent, has been used for the treatment of GC patients, which is confirmed to result in DNA damage, leading to the death of the rapidly proliferating cells. 4 However, cisplatin resistance reduced improvement in the survival of cisplatin treated GC patients. Thus, it is urgent to find new methods to alleviate cisplatin resistance in GC.
Long noncoding RNAs have been identified to regulate transcriptional and post-transcriptional gene expression. Long noncoding RNA regulator of reprogramming (ROR) has been demonstrated to be dysregulated in various types of cancers, including papillary thyroid carcinoma, 5 breast cancer, 6 hepatocellular carcinoma, 7 glioblastoma cancer, 8 esophageal squamous cell carcinoma, 9 and also in GC. 10 ROR was demonstrated to be involved in the tamoxifen resistance through sponging miR-205 in breast cancer. 11 However, the biological mechanism of ROR was still undefined in cisplatin resistance of GC.
MicroRNAs (miRNAs) have been documented to function as gene expression inhibitor through inhibiting message RNAs (mRNAs) translation or mediating target mRNAs degradation. Accumulating evidence raveled that miR-519d-3p was dysregulated in many cancers, including breast cancer, 12 pancreatic cancer, 13 and GC. 14 High mobility group protein A2 (HMGA2) is a DNA binding protein, which assembles at the enhancer and regulates expression of the target gene. 15 HMGA2 was reported to be abnormally expressed in many human cancers, including pituitary neuroendocrine tumors, 16 glioma, 17 breast cancer, 18 and GC. 19,20 However, the effects and biological mechanism of miR-519d-3p and HMGA2 remain unclear in GC.
In our research, we validated that ROR and HMGA2 expression were increased, and miR-519d-3p was downregulated in GC tissues and cells. Moreover, we first established the novel regulatory network ROR/miR-519d-3p/HMGA2 in cisplatin resistance of GC, and this may provide basis for the further GC study.
Materials and Methods
Tissue sample
Thirty GC tissues and their corresponding adjacent normal tissues were obtained from the People's Hospital of Zhangqiu. The clinicopathologic features of these patients are presented in Table 1. The study was approved by the Ethics Committee of the People's Hospital of Zhangqiu, and GC patients provided written informed consents.
Correlation of the Expression of Regulator of Reprogramming with Clinicopathologic Features in Patients of Gastric Cancer
ROR, regulator of reprogramming.
Cell culture and transfection
MKN45 and HGC-27 cell lines, as well as human gastric epithelial cell line (GES-1) (China center for type culture collection, Wuhan, China), were cultivated in RPMI-1640 medium (BIOSUN, Shanghai, China) containing 10% fetal bovine serum (FBS; Thermo Fisher Scientific, Rockville, MD) in an incubator with 37°C and 5% CO2. GC cells were selected by treating with 0.01–100 μM cisplatin for 72 h. Short hairpin RNA (shRNA) targeted for ROR (sh-ROR, GGAGAGGAAGCCUGAGAGU) and its negative control (sh-NC), miR-519d-3p inhibitor (anti-miR-519d-3p) and its mock (anti-miR-NC), miR-519d-3p mimics (miR-519d-3p) and its mock (miR-NC), ROR overexpression vector pcDNA-ROR (ROR), HMGA2 overexpression vector pcDNA-HMGA2 (HMGA2) and empty vector pcDNA (vector) purchased from GenePharma (Shanghai, China) were transfected into GC cells by Lipo-fectamine 2000 Reagent (Invitrogen, Carlsbad, CA).
Quantitative real-time polymerase chain reaction
RNA was extracted using Trizol reagent (Invitrogen) and reversed into cDNA. RNA was quantified using a NaniDrop ND-1000 Spectrophotometer (NaniDrop, Wilmington, MA), and its purity was detected using the A260/280 ratio. Then, qRT-PCR was performed using Real-Time PCR Detection System (Bio-Rad, Shanghai, China), and the data analyzed by the 2−ΔΔCt method. The primers were as follows: ROR: CGAACGAGAGGACCGAAG (sense) and GCCAAGTTCTAGATAAGC (antisense); miR-519d-3p: TGCGGCAAAGTGCCTCCCTTTAG (sense) and CCAGTGCAGGGTCCGAGGT (antisense); HMGA2: AAAGCAGCTCAAAAGAAAGCA (sense) and TGTTGTGGCCATTTCCTAGGT (antisense); U6: CTCGCTTCGGCAGCACA (sense) and AACGCTTCACGAATTTGCGT (antisense); GAPDH: TGTTCGTCATGGGTGTGAAC (sense) and ATGGCATGGACTGTGGTCAT (antisense).
Western blot
RIPA Lysis (Thermo Fisher Scientific) was used for protein extraction from cells. Then SDS-PAGE was used to separate proteins, and the separated protein was transferred onto PVDF membrane and blocked, and then incubated with the primary antibody, the secondary antibody in order. Clarity™ Western ECL Substrate Kit (Bio-Rad) was utilized to detect the chemiluminescence intensity. All the antibodies used in the present research were obtained from Abcam (Cambridge, MA).
3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT; Promega, Madison, WI) assay
Cells were cultured in a 96-well plate for 0, 24, 48, and 72 h. Then the resuspended cells were incubated with 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) for 3 h; dimethyl sulfoxide (DMSO) was added into the well to dissolve the formazan for 15 min at 37°C. A spectrophotometer (Thermo Fisher Scientific) was used to determine the absorbance at 570 nm.
Transwell assay
The migration assay was performed according to its manual (Corning, Tewksbury, MA). In brief, cells cultured with serum-free medium were plated in the upper chamber, while medium containing 10% FBS was used in the lower chamber. After incubation, cells in the bottom were fixed, stained, and counted. Four fields were randomly selected, and a light microscope (Olympus, Japan) was used to count the cell numbers. For the invasion assay, the protocol was modified such that the upper chamber was precoated with Matrigel (BD Biosciences, San Jose, CA).
Flow cytometry
For the cell apoptosis assay, the cells were treated with annexin V-fluorescein isothiocyanate (FITC) (10 μL) and propidium iodide (PI) (10 μL) (Sigma-Aldrich, St. Louis, MO) to differentiate apoptotic cells and viable or necrotic cells. Apoptotic cells were analyzed by flow cytometry.
Measurement of caspase3 activity
Total protein was isolated from GC cells. Caspase-3 Colorimetric Assay kit (Nanjing KeyGen Biotech Co., Ltd., China) was used to determine the caspase3 activity according to the manufacturer's protocol.
Dual luciferase reporter assay
The putative target of ROR was predicted by miRcode, and the putative target of HMGA2 was predicted by starBase v2.0 online database. The sequences of ROR and HMGA2 3′UTR were amplified and inserted into psiCHECK2 vector (Promega), named WT-ROR and HMGA2 3′UTR-WT, respectively. The luciferase reporter WT-ROR (MUT-ROR) or HMGA2 3′UTR-WT (HMGA2 3′UTR-MUT) and miR-519d-3p mimics (miR-NC) were cotransfected into MKN45 and HGC-27 cells using a dual luciferase reporter assay kit (Promega). The Renilla luciferase activities were used to normalize the firefly luciferase activities.
RNA immunoprecipitation assay
Magna RNA immunoprecipitation (RIP)-Kit (Millipore, Bedford, MA) was used in RIP assay. MKN45 and HGC-27 cells were lysed, and incubated with anti-Ago2 or anti-IgG control (Abcam), and then cultured with protein A magnetic beads. Coprecipitated RNA used for quantitative real-time polymerase chain reaction (qRT-PCR) was bound to primary Ago2 antibody.
In vivo experiment
This experiment was performed in accordance with the manual approved by the Animal Care Committee of the People's Hospital of Zhangqiu. Six-week-old female BALB/c athymic nude mice (n = 7 per group) were purchased from Shanghai Laboratory Animal Center (Shanghai, China). MKN45 cells transfected with sh-ROR or sh-NC were subcutaneously injected into the mice. The tumor volumes were calculated every 4 d for 4 weeks after injection. The tumor samples were excised from mice, which were raised for 28 d after inoculation for weight measurement and for the subsequent qRT-PCR and Western blot analysis.
Statistical analysis
The data were represented as mean ± standard deviation. The differences between two and more than three groups were analyzed by Student's t-test and analysis of variance, respectively. Pearson's correlation analysis was used to analyze the relationship between ROR and miR-519d-3p or HMGA2. p < 0.05 was regarded as statistically significant difference.
Results
ROR and HMGA2 are significantly upregulated in GC tissues
To explore the role of ROR and HMGA2 in GC, we first measured the level of ROR and HMGA2 in GC tissues by qRT-PCR. The results showed that the levels of ROR and HMGA2 were significantly increased in GC tissues compared with those in the corresponding adjacent tissues (Fig. 1A, B). In addition, the scatter diagram displayed that the level of HMGA2 was positively correlated with the level of ROR (Fig. 1C). Western blot results presented that the protein level of HMGA2 was highly expressed in GC tissues (Fig. 1D). Immunohistochemical analysis results showed that HMGA2 was highly expressed in GC tissues (Fig. 1E). Taken together, the levels of ROR and HMGA2 were both apparently increased in GC tissues.

ROR and HMGA2 were significantly upregulated in GC tissues. The levels of ROR
ROR knockdown inhibits cell proliferation, metastasis, and cisplatin resistance in GC cells
To examine the effect of ROR in GC, ROR was depleted in this study. First, the qRT-PCR results indicated that ROR was also greatly elevated in MKN45 and HGC-27 cells in contrast to those in GES-1 (Fig. 2A). Subsequently, the qRT-PCR results verified knockdown efficiency indicated by the conspicuous downregulation of ROR in sh-ROR-transfected MKN45 and HGC-27 cells (Fig. 2B). Furthermore, the MTT and transwell assays showed that cell viability, migration and invasion were all remarkably reduced in MKN45 and HGC-27 cells transfected with sh-ROR related to that in si-NC group (Fig. 2C–F). Since E-cadherin, N-cadherin, and Vimentin were reported as marker proteins in epithelial–mesenchymal transition (EMT), 21 we detected the effects of ROR depletion on the protein levels of E-cadherin, N-cadherin, and Vimentin. Western blot assay exhibited that E-cadherin was distinctly upregulated in sh-ROR-transfected MKN45 and HGC-27 cells compared with those in si-NC treatment, while N-cadherin and Vimentin were both notably decreased in MKN45 and HGC-27 cells transfected with sh-ROR (Fig. 2G, H). To explore whether ROR was related to cisplatin resistance in GC, MKN45 and HGC-27 cells were treated with 0.01–100 μM cisplatin for 72 h. Our results indicated that downregulation of ROR decreased the half-maximal inhibitory concentration (IC50) of GC cells treated with cisplatin (Fig. 2I, J). Moreover, ROR deletion could increase the number of apoptotic cells and the expression of caspase3 (Fig. 2K, L). These results manifested that ROR silencing suppressed cell proliferation, migration, invasion, EMT and enhanced sensitivity of GC cells to cisplatin in GC cells.
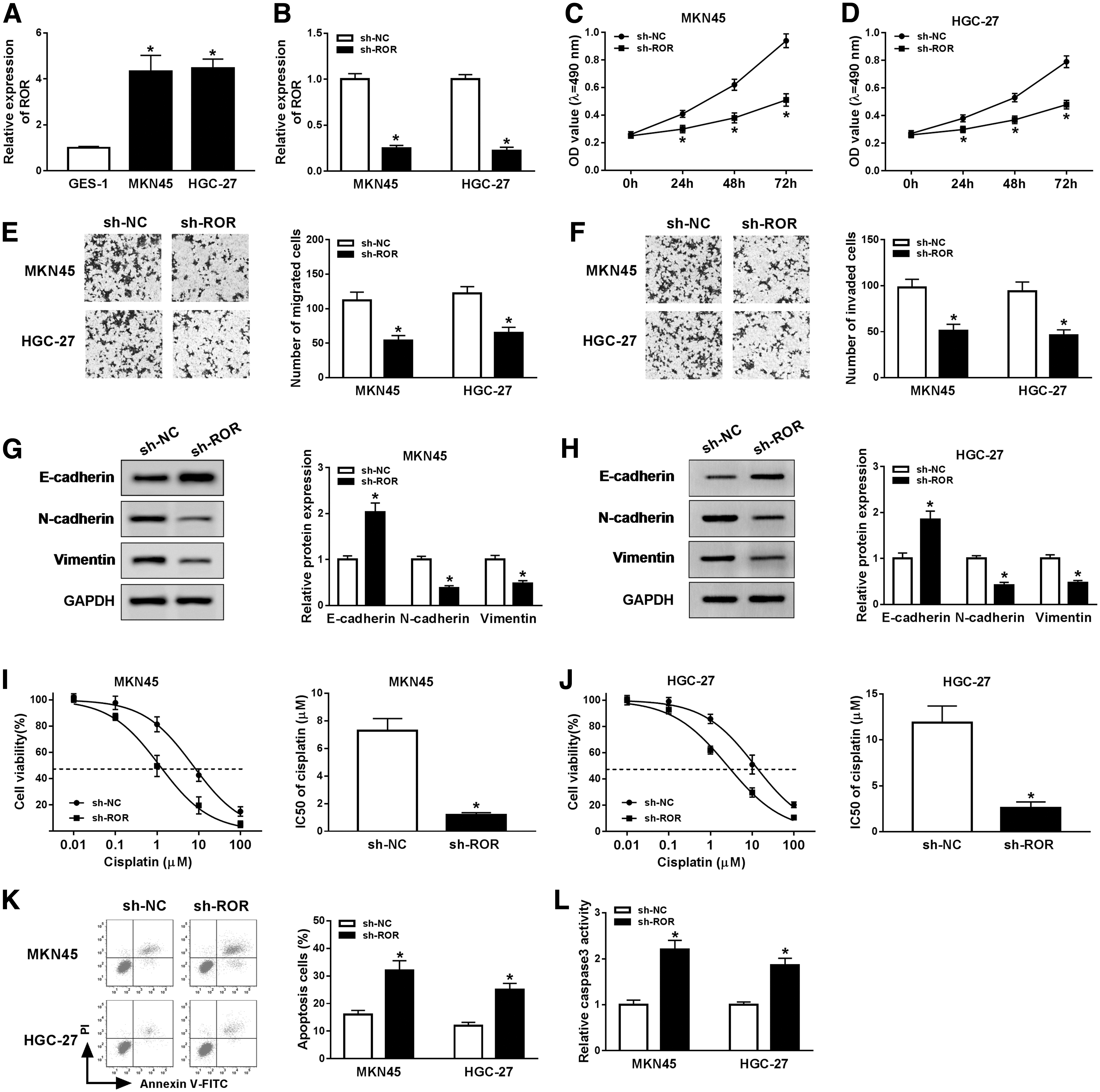
ROR knockdown inhibited cell proliferation, migration, invasion, EMT, and cisplatin resistance in GC cells. The level of ROR in MKN45 and HGC-27 cells
HMGA2 overexpression could reverse ROR knockdown-induced inhibitory effects rather than cell proliferation, migration, invasion, and cisplatin resistance
To further explore the relationship between ROR and HMGA2 in GC, we first assessed the protein level of HMGA2 in MKN45 and HGC-27 cells. The Western blot assay displayed that HMGA2 was markedly upregulated in GC cells (Fig. 3A). As shown in Figure 3B, HMGA2 showed the successful overexpression efficiency in both MKN45 and HGC-27 cells. qRT-PCR confirmed the successful overexpression efficiency of ROR in both MKN45 and HGC-27 cells (Fig. 3C). Also, the protein level of HMGA2 in MKN45 and HGC-27 cells was evidently increased in ROR overexpression group compared with that in its matched control, while the transfection of sh-ROR resulted in the downregulation of HMGA2 expression (Fig. 3D). Moreover, the MTT assay and transwell assay showed that the cell viability, migrated cells, and invaded cells were all strikingly downregulated in MKN45 and HGC-27 cells transfected with sh-ROR, while overexpression of HMGA2 attenuated the inhibitory effects of ROR deletion on cell viability, migrated cells, and invaded cells (Fig. 3E–H). In addition, the Western blot assay exhibited that HMGA2 overexpression reversed the promotion effect of ROR deletion on E-cadherin and inhibition effects of ROR deletion on N-cadherin and Vimentin expression in MKN45 and HGC-27 cells (Fig. 3I, J). Our data also suggested that ROR deletion enhanced cisplatin sensitivity, as shown by decreased IC50s and upregulated numbers of apoptotic cells and caspase3 expression, which was reversed by upregulation of HMGA2 (Fig. 3K–M). To sum, these data demonstrated that HMGA2 overexpression mitigated the inhibitory effects on cell proliferation, migration, invasion, EMT, and cisplatin resistance in GC cells induced by ROR knockdown.
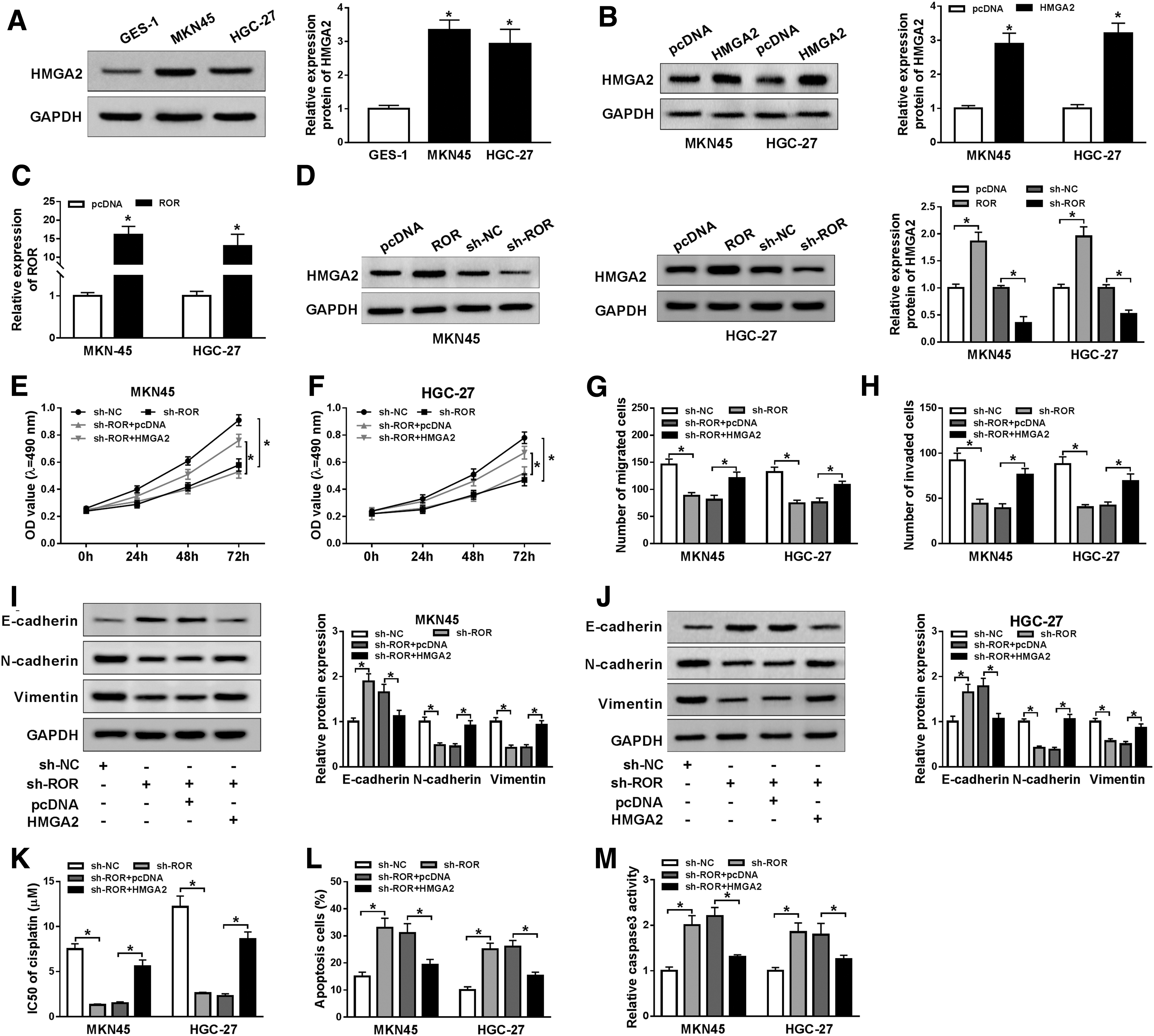
HMGA2 overexpression alleviated the inhibitory effects on cell proliferation, migration, invasion, and cisplatin resistance in GC cells induced by ROR knockdown.
ROR regulates HMGA2 through sponging miR-519d-3p in GC cells
To investigate the biological relationship among ROR, miR-519d-3p, and HMGA2 in GC, miRcode and starBase v2.0 online databases were used to predict the putative target of ROR and HMGA2, respectively. The results showed that miR-519d-3p had the complementary binding sites with ROR and HMGA2 (Fig. 4A, B). Then, we determined the overexpression efficiency of miR-519d-3p and found the successful overexpression efficiency in both MKN45 and HGC-27 cells (Fig. 4C). Dual luciferase reporter assay indicated that miR-519d-3p mimics inhibited the luciferase activity in WT-ROR group, while no significant change was observed in MUT-ROR group (Fig. 4D, E). Also, the dual luciferase reporter assay showed the same trend between miR-519d-3p and HMGA2 3′UTR (Fig. 4F, G). RIP assay revealed that ROR, miR-519d-3p, and HMGA2 were precipitated by Anti-Ago2, indicating the coexpression of ROR, miR-519d-3p, and HMGA2 in GC (Fig. 4H, I). Furthermore, the Western blot results exhibited that the protein level of HMGA2 was strikingly reduced in MKN45 and HGC-27 cells transfected with miR-519d-3p, while upregulation of ROR mitigated the inhibitory effect (Fig. 4J, K). These data unraveled that miR-519d-3p negatively interacted with ROR and HMGA2, and ROR regulated HMGA2 expression by regulating miR-519d-3p in GC cells.
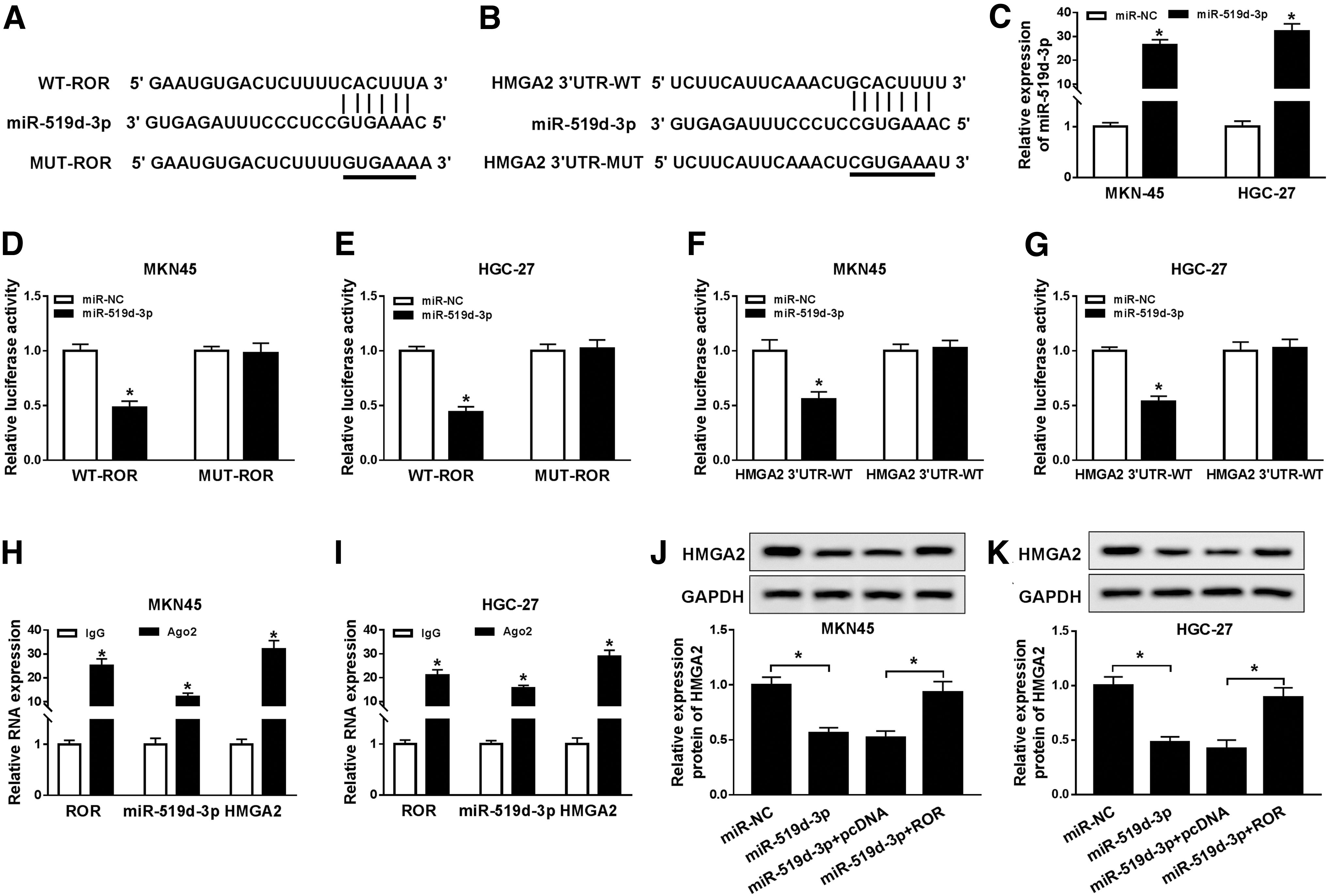
ROR regulated HMGA2 through sponging miR-519d-3p in GC cells. The putative complementary binding sites between miR-519d-3p and ROR
miR-519d-3p inhibitor blocks the inhibitory effects on cell proliferation, migration, invasion, EMT, and cisplatin resistance in GC cells caused by ROR silencing
To evaluate the role of miR-519d-3p in GC, the level of miR-519d-3p was first measured in GC tissues and cells. The qRT-PCR results exhibited that the level of miR-519d-3p was obviously downregulated in GC tissues and cells (Fig. 5A, C). As shown in Figure 5D, anti-miR-519d-3p showed the successful knockdown efficiency in both MKN45 and HGC-27 cells. The level of miR-519d-3p was negatively linearly correlated with the level of ROR (Fig. 5B). The MTT assay, transwell assay, and Western blot assay indicated that anti-miR-519d-3p mitigated the inhibitory effects on cell viability, migrated cells, invaded cells, and the protein levels of N-cadherin, Vimentin induced by sh-ROR, while the protein level of E-cadherin showed the opposite trends (Fig. 5E–J). Moreover, we found that ROR deletion decreased IC50s, and upregulated numbers of apoptotic cells and caspase3 expression, thereby enhanced cisplatin sensitivity, which was reversed by downregulation of miR-519d-3p (Fig. 5K–M). These data revealed that miR-519d-3p inhibitor alleviated the inhibitory effects on cell proliferation, migration, invasion, EMT, and cisplatin resistance in GC cells induced by ROR depleted.
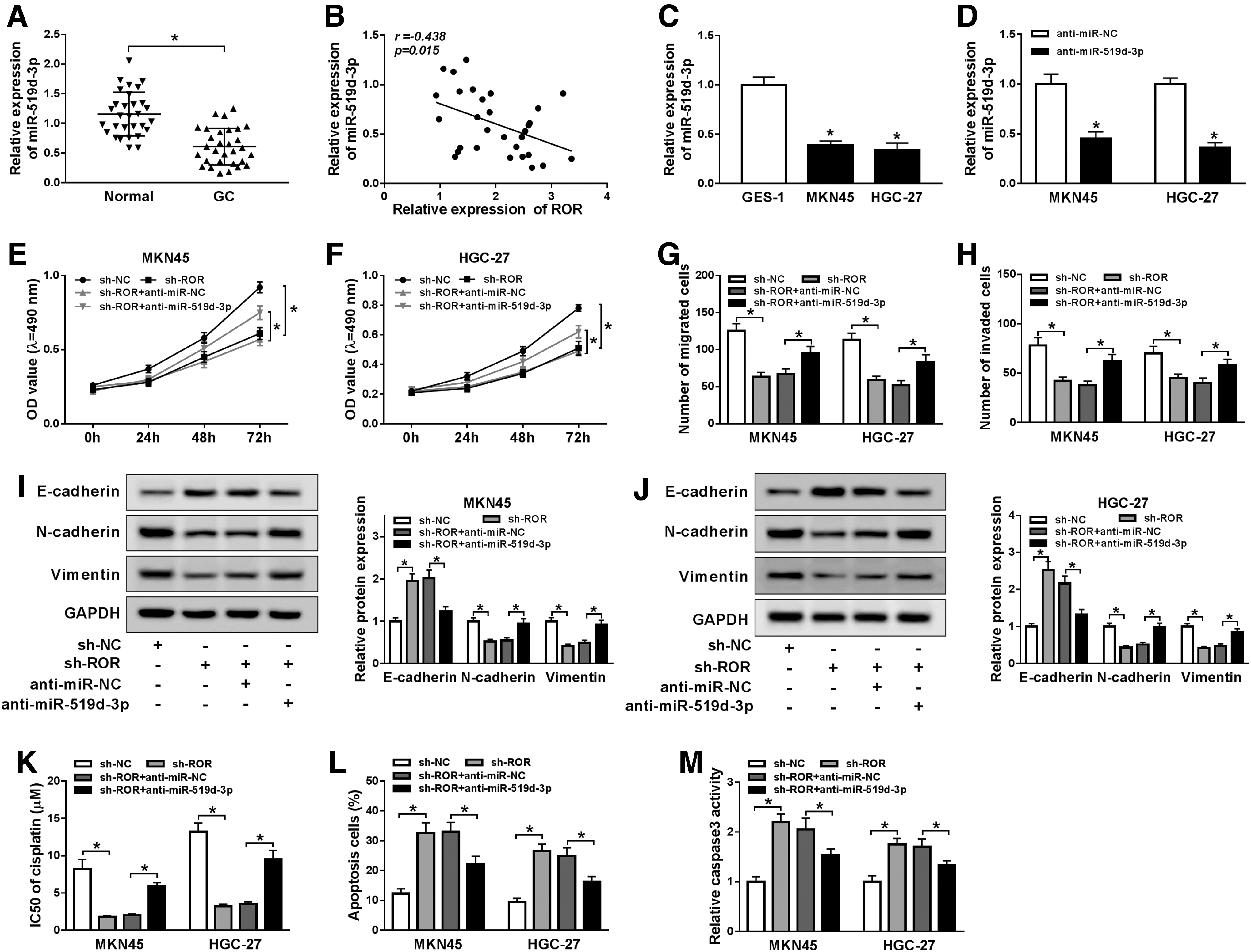
miR-519d-3p inhibitor blocked the inhibitory effects on cell proliferation, migration, invasion, EMT, and cisplatin resistance in GC cells caused by ROR silencing. The level of miR-519d-3p in GC tissues (n = 30)
ROR knockdown decreases HMGA2 to restrain xenograft tumor growth and EMT by upregulating miR-519d-3p in vivo
To confirm the functional role of ROR in vivo, GC cells transfected with sh-ROR or sh-NC were subcutaneously injected into nude mice. After the 4 weeks measurement, the tumor volume and weight were both significantly decreased in sh-ROR group (Fig. 6A, B). The qRT-PCR results indicated that the level of ROR was apparently downregulated, and miR-519d-3p was dramatically upregulated in sh-ROR group in comparison with that in sh-NC group (Fig. 6C). In addition, the protein level of E-cadherin was remarkably increased, and HMGA2, N-cadherin, and Vimentin were notably decreased in sh-ROR group (Fig. 6D). These results manifested that ROR silencing reduced HMGA2 expression to suppress xenograft tumor growth and EMT by upregulating miR-519d-3p in vivo.

ROR knockdown restrained xenograft tumor growth and EMT by upregulating miR-519d-3p in vivo. Tumor volume
Discussion
In this study, we focused on the biological mechanism of ROR in GC. All results in this project unraveled that lncRNA ROR regulate cell growth, metastasis, and cisplatin resistance through miR-519d-3p/HMGA2 axis in GC.
ROR was proved to be associated with cancer progression and transition in various types of cancers, including GC. ROR was confirmed to play vital role in cancer resistance. For instance, a previous study on breast cancer documented that ROR was significantly increased in tumor tissues; ROR silencing restrained cell growth and upregulated sensitivity to tamoxifen in breast cancer. 22 Li et al. reported that ROR overexpression reduced gemcitabine sensitivity in pancreatic cancer cells. 23 In this study, we demonstrated that ROR exhibited a high expression in GC tissues and cells. Moreover, we analyzed the correlation of ROR expression with clinicopathologic features in GC patients. Clinical data indicated that ROR expression was closely associated with stages or lymph node metastasis. Furthermore, ROR silencing apparently inhibited cell viability, migrated cells, invaded cells, and EMT process, as well as cisplatin resistance in MKN45 and HGC-27 cells. In addition, ROR deletion restrained tumor growth in vivo.
Emerging evidence manifested that HMGA2 was abnormally expressed in many human cancers, including GC. For example, HMGA2 expression was higher in GC tissues and exerted promotion effects in GC, and HMGA2 knockdown inhibited cell metastasis in GC cells. 19,20 HMGA2 participated in drug resistance of various cancers. 24,25 Moreover, Dong et al. reported that HMGA2 played a promotion role in GC cell metastasis. 26 Another study illustrated that HMGA2 could elevate EMT in GC through TWIST1. 27 More importantly, HMGA2 might be a prognosis biomarker in GC. 28 In the present research, our data indicated that HMGA2 was dramatically enhanced in GC tissues and cells. HMGA2 harbored the inhibition effects on cell viability, migrated cells, invaded cells, and EMT, as well as cisplatin resistance induced by sh-ROR in GC cells.
MiR-519d-3p was dysregulated and affected tumor progression in many cancers, including GC. Previous studies reported that miR-519d-3p regulated cell proliferation, migration, invasion, and EMT in GC cells. 14,29 In this study, miR-519d-3p was markedly downregulated in GC tissues and cells. Moreover, miR-519d-3p directly interacted with ROR and HMGA2. Further, miR-519d-3p inhibitor attenuated the inhibitory effects on cell viability, migrated cells, invaded cells, and EMT, as well as cisplatin resistance induced by ROR silencing in GC cells. In addition, ROR knockdown modulated HMGA2 expression by sponging miR-519d-3p.
Conclusions
In summary, we first reported that ROR modulated HMGA2 to regulate cell proliferation, migration, invasion, EMT, and cisplatin sensitivity by sponging miR-519d-3p in GC, and ROR/miR-519d-3p/HMGA2 regulatory network may provide basis for further GC study.
Footnotes
Authors' Contributions
W.J. and S.L. performed conception and design; H.Z. contributed to development of methodology; M.L. performed acquisition, analysis, and interpretation of data; W.J., H.Z., and S.L. contributed to writing, review, and revision of the article; all coauthors have reviewed and approved the article before submission.
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.