
Abstract
Background:
Hepatocellular carcinoma (HCC) is a common malignancy worldwide. Radiofrequency ablation (RFA) is applied for treating HCC; however, insufficient RFA promotes HCC development and accelerates HCC recurrence. Therefore, the molecular functions underlying this process have gradually attracted attention.
Objective:
We sought to examine whether GAS6-AS2 (also known as GAS6-DT: growth arrest specific 6 divergent transcript) played a role in the development of HCC after insufficient RFA.
Materials and Methods:
The in vitro model was established by heating Huh7 and MHCC97 cells in water bath at 47°C, named as Huh7-H and MHCC97-H. Colony formation, transwell and Western blot assays were conducted for functional analysis.
Results:
GAS6-AS2 was upregulated in Huh7-H and MHCC97-H cells relative to Huh7 and MHCC97 cells. GAS6-AS2 deficiency hampered cell proliferation, migration, invasion, epithelial-mesenchymal transition, and stemness in Huh7-H and MHCC97-H cells. Moreover, microRNA-3619-5p (miR-3619-5p) combined with GAS6-AS2 and ARL2 (ADP ribosylation factor-like GTPase 2) was the target gene of miR-3619-5p. GAS6-AS2 served as the competing endogenous RNA (ceRNA) of ARL2 via absorbing miR-3619-5p.
Conclusion:
On the whole, present study uncovered a novel ceRNA mechanism of GAS6-AS2/miR-3619-5p/ARL2 in HCC after insufficient RFA, which might shed a new insight into treatment of HCC after insufficient RFA.
Introduction
Hepatocellular carcinoma (HCC) is a common malignancy with high morbidity and mortality worldwide. 1 HCC is prevalent in China and is the third common cause of cancer deaths in 2015. 1 Despite the fact that liver transplantation is the most preferred treatment for patients with HCC, its application is limited by short supply of suitable donor organs. Hence, radiofrequency ablation (RFA) is applied for another option when treating HCC.
Presently, RFA is widely used for HCC patients who cannot undergo surgical resection or liver transplantation. However, it is difficult for HCC to achieve sufficient ablation due to multiple factors, such as anatomical location restrictions, insufficient ablative margin, and heat loss, in ablation for a large tumor. 2,3 More seriously, HCC after insufficient RFA is prone to more rapid and aggressive recurrence. Thus, it is of great significance to explore the mechanism underlying the initiation and development of HCC with insufficient RFA.
Long noncoding RNAs (lncRNAs) are a group of noncoding RNAs (ncRNAs) longer than 200 nucleotides. It was widely reported that lncRNAs play a crucial part in HCC. LncRNA ID2-AS1 blocks HCC progression by activation of HDAC8/ID2 pathway. 4 LncRNA 91H contributes to HCC growth and migration through its epigenetic regulation on IGF2 expression. 5 Insufficient RFA was unmasked to promote cancer progression. For instance, insufficient RFA promotes nonsmall cell lung cancer cell growth via activating PI3K/Akt/HIF-1α pathway. 6 Insufficient RFA promotes cell proliferation and angiogenesis in residual lung carcinoma through activation of HSP70/HIF-1α signals. 7 Also, some reports revealed that lncRNAs are involved in HCC development after insufficient RFA in cancers. For instance, silenced lncRNA FUNDC2P4 facilitates epithelial–mesenchymal transition (EMT) in residual HCC after insufficient RFA. 8 STIP1 mediates HCC metastasis after insufficient RFA. 9
LncRNA GAS6-AS2 (GAS6 divergent transcript, also known as GAS6-DT) is verified to accelerate cell proliferation and hinder apoptosis in melanoma via elevating GAS6 expression. 10 Also, GAS6-AS2 facilitates bladder cancer development via miR-298/CDK9 axis. 11 Present study was designed to reveal the role and possible mechanism of GAS6-AS2 in HCC under insufficient RFA.
Materials and Methods
Cell culture
Human hepatocellular carcinoma cell lines (HuH7 and MHCC97) and the human normal live cells (THLE-2) were procured from Chinese Academy of Sciences (Shanghai, China). Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco, Grand Island, NY) was supplied with 1% mixture of penicillin/streptomycin and 10% fetal bovine serum for cell culture purposes under 37°C and 5% CO2.
Establishment of in vitro insufficient RFA cell model
Cells (1 × 105) were seeded into the 7 cm diameter Petri dishes for 24 h of culture. Cells were treated with the repeated cycles as below: cells on parafilm sealed plates were submerged for 5 min into the 47°C water bath, recovered under 37°C with 5% CO2, and heated sequentially at about 80% confluent in water bath at 47°C for 10, 15, 20, and 25 min in each cycle. The survived cells collected from the last cycle were separately named as Huh7-H and MHCC97-H.
Extraction of total RNA and quantitative real-time polymerase chain reaction
Using TRIzol reagent, the total RNA was extracted from cultured cell lines as requested of protocol (Invitrogen, Carlsbad, CA). Following identifying RNA concentration and quality, synthesized cDNA was obtained by reverse transcription with PrimeScript RT Reagent Kit (TaKaRa, Tokyo, Japan). Afterward, SYBR Premix Ex Taq II kit (TaKaRa) was applied for quantitative real-time polymerase chain reaction (qRT-PCR). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) or U6 served as the endogenous normalizer. Relative gene expression was tested by 2−ΔΔCt method.
Plasmid transfection
Cells were planted in 24-well culture plates for 48 h of plasmid transfection with the application of Lipofectamine2000 (Invitrogen). All of the transfection plasmids, including the designed short hairpin RNAs (shRNAs) for GAS6-AS2 and the control sh-NC, miR-3619-5p mimics/inhibitor, and NC mimics/inhibitor, as well as pcDNA3.1/ARL2 and the empty pcDNA3.1 control, were procured from Genepharma Company (Shanghai, China). ShRNA sequences were listed as below: sh-NC: 5′-CCGGTTGGATGAAAATTTTTGCTTTCTCGAGAAAGCAATTTTTAACATGGTCTTTTTG-3 ′; sh-GAS6-AS2#1: 5′-CCGGGAGCATGTTAATTTAAGCAAACTCGAGTTTGCTTAAATTAACATGCTCTTTTTG-3′; sh-GAS6-AS2#2: 5′-CCGGATGATCTTCAAGGAGAATTAACTCGAGTTAATTCTCCTTGAAGATCATTTTTTG-3′.
Colony formation
The processed cells were planted into six-well plates and cultivated at 37°C with 5% CO2 for 14 d. After removing culture medium, cells were rinsed twice in phosphate-buffered saline (PBS; Invitrogen). One milliliter of 0.1% crystal violet solution was then added into each well. At length, colonies number was counted manually. The experiment was repeated three times.
Transwell assay
Cell cultured in serum-free medium were seeded in the upper chamber of 8 mm pore size Transwell chambers (Corning, Corning, NY) to assess cell migration. Cell invasion was examined using the Matrigel-coated transwell chamber. The lower chamber was supplied with the 100% complete medium. Twenty-four hours later, cells on the bottom were counted under microscopy (Olympus, Tokyo, Japan) after fixed and stained. The experiment was repeated three times.
Sphere formation assay
Sphere formation assay was operated to assess the spheroid-formation ability of indicated HCC cells. In short, cells suspension in RPMI 1640 medium containing epidermal growth factor (20 ng/mL; Promega), basic fibroblast growth factor (10 ng/mL; Promega), and 2% B27 (Gibco, Thermo Fisher Scientific) were supplemented into in 6-well plates with ultralow adhesion (Corning). After culturing for 1–2 weeks, the spheres were observed and those larger than 100 μm were counted by using a confocal microscope (Leica, Germany).
Western blot
After culturing in Radio Immunoprecipitation Assay buffer, cell lysates were first separated on 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membrane. After blocking, membranes were cultured with primary antibodies at 4°C overnight and then with horseradish peroxidase-conjugated secondary antibody. Antibodies against GAPDH (loading control; ab181602, 1:10,000) and N-cadherin (ab18203, 1:1000), E-cadherin (ab40772, 1:10,000), Nanog (ab109250, 1:2000), OCT4 (ab27985, 1:1000), and ARL2 (ab183510, 1:2000) were acquired from Abcam (Cambridge, MA). Protein signals were detected by ECL method (GE Healthcare, Chicago, IL). The experiment was repeated three times.
Subcellular fraction assay
Huh7-H and MHCC97-H cells were washed twice in PBS. Cell lysates from cell fractionation buffer were centrifuged to isolate nucleus and cytoplasm. The supernatant was seen as cytoplasmic fraction. The remaining lysates were washed, centrifuged, and treated with cell disruption buffer. The relative levels of GAS6-AS2, nuclear control U6, and cytoplasmic control GAPDH were detected by qRT-PCR. The experiment was repeated three times.
RNA immunoprecipitation
By use of the Magna RIPTM RNA-Binding Protein Immunoprecipitation Kit, RNA immunoprecipitation (RIP) assay was implemented as requested by supplier (Millipore, Bedford, MA). Magnetic beads were precoated with human Ago2 antibody or normal control immunoglobulin G (IgG) antibody for immunoprecipitation with cell lysates in RIP buffer. qRT-PCR was conducted for the quantification of RNAs in precipitates. The experiment was repeated three times.
RNA pull down
The GAS6-AS2 and ARL2 target sequences (wild type or mutated) within miR-3619-5p fragments were biotinylated into Bio-miR-3619-5p-WT/Mut. Cell extracts were mixed with magnetic beads and above labeled RNAs (with Bio-NC as the negative control). The pulled-down RNAs were quantitated and tested by qRT-PCR.
Luciferase reporter assay
The miR-3619-5p target sites (wild type or mutated) within GAS6-AS2 fragment and ARL2 3′UTR (3′untranslated region) fragment were inserted into pmirGLO Dual-Luciferase Expression Vector (Promega Corporation, Fitchburg, WI). The constructed GAS6-AS2-WT/Mut or ARL2 3′UTR-WT/Mut reporter vectors were cotransfected with miR-3619-5p mimics or NC mimics. Dual-Luciferase Reporter Assay System (Promega) was utilized to assess luciferase activity at 48 h posttransfection. The experiment was repeated three times.
Statistical analysis
All experimental data are exhibited as mean ± standard deviation and analyzed by GraphPad PRISM 6 (GraphPad, San Diego, CA). Variance between two groups or among three groups was assayed by Student's t test and one-way analysis of variance, respectively. The significance threshold was set as p < 0.05. Each assay was repeated thrice.
Results
GAS6-AS2 promoted cell proliferation, migration, invasion, EMT, and stemness
To evaluate proliferation changes in Huh7-H and MHCC97 cells, the colony formation assay was carried out. Huh7-H and MHCC97-H cells possessed stronger proliferation ability than the parental Huh7 and MHCC97 cells (Supplementary Fig. S1A). By means of transwell assay, heating HCC cells acquired facilitated migration and invasion ability compared to the parental control cells (Supplementary Fig. S1B). Besides, the authors also disclosed that heating treatment made HCC cells equipped with enforced EMT ability and stemness characteristic (Supplementary Fig. S1C, D). Moreover, the sphere formation ability of both Huh7 and MHCC97 cells was also strengthened after heating treatment (Supplementary Fig. S1E). Then, they detected the expression of GAS6-AS2 in Huh7, MHCC97, and normal THLE-2 cells. Result revealed that GAS6-AS2 was remarkably upregulated in malignant Huh7 and MHCC97 cells in comparison to THLE-2 cells, and its expression was further elevated in cancerous cells by water bathing (Fig. 1A). Next, they silenced GAS6-AS2 in Huh7-H and MHCC97-H cells, with the knockdown efficiency verified by qRT-PCR (Fig. 1B). It was disclosed that GAS6-AS2 depletion reduced cell proliferation ability in both Huh7-H and MHCC97-H cells (Fig. 1C). Furthermore, cell migration and invasion abilities were both hindered after GAS6-AS2 downregulation (Fig. 1D). Besides, inhibition of GAS6-AS2 restrained EMT process and stemness in these two cells (Fig. 1E, F, and Supplementary Fig. S2A, B). In a word, silencing GAS6-AS2 obstructed cell proliferation, migration, invasion, EMT, and stemness in Huh7-H and MHCC97-H cells.
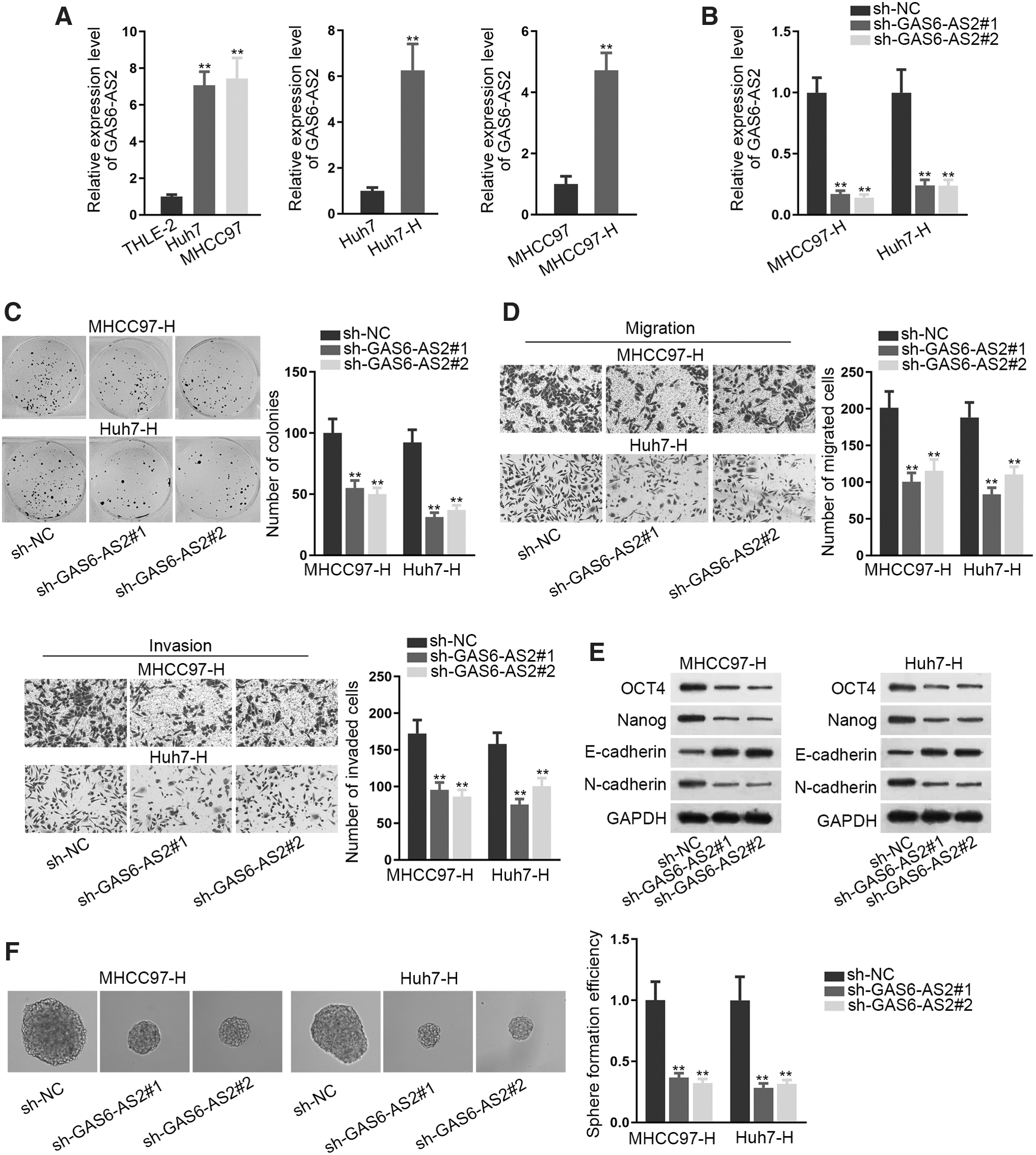
GAS6-AS2 promoted cell proliferation, migration, invasion, EMT, and stemness.
MiR-3619-5p was sponged by GAS6-AS2
Subsequently, the subcellular location of GAS6-AS2 was determined by subcellular fraction assay and the result revealed GAS6-AS2 as a cytoplasmic RNA (Fig. 2A). Next, RIP assay revealed that GAS6-AS2 was abundantly enriched in anti-Ago2 (Fig. 2B), which indicated that GAS6-AS2 was involved in RNA-induced silencing complexes (RISCs) and might serve as a competing endogenous RNA (ceRNA) in Huh7-H and MHCC97-H cells. Based on ENCORI database, 12 eight candidate miRNAs were identified. Among them, only miR-3619-5p was dramatically downregulated in Huh7-H and MHCC97-H cells (Fig. 2C). The putative binding sites between GAS6-AS2 and miR-3619-5p were predicted from ENCORI database, while the mutated GAS6-AS2 sequences that could not be recognized by miR-3619-5p were also presented (Fig. 2D). It was demonstrated that GAS6-AS2 was remarkably pulled down by biotinylated miR-3619-5p-WT, but not by biotinylated miR-3619-5p-Mut (Fig. 2E). Besides, under enhanced expression of miR-3619-5p in Huh7-H and MHCC97-H cells (Fig. 2F), the luciferase activity of wild GAS6-AS2-WT was reduced while that of GAS6-AS2-Mut not impacted (Fig. 2G). Furthermore, it was demonstrated that elevating miR-3619-5p expression in Huh7-H and MHCC97-H cells suppressed cell proliferation, migration, invasion, EMT, and stemness (Fig. 2H–J and Supplementary Fig. S2C–E). In brief, GAS6-AS2 interacted with miR-3619-5p, which served an antitumor part in Huh7-H and MHCC97-H cells.
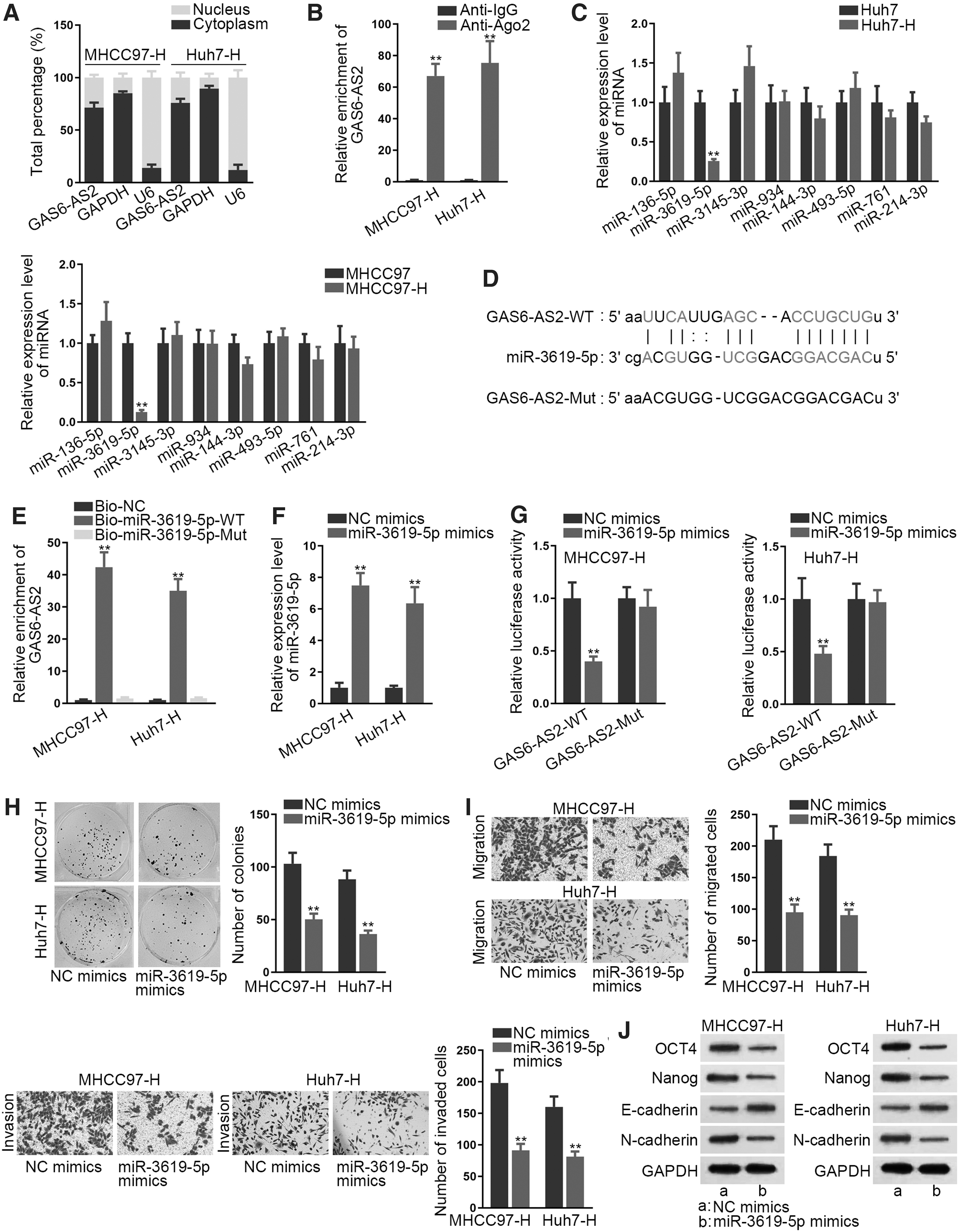
MiR-3619-5p was sponged by GAS6-AS2.
ARL2 was targeted by miR-3619-5p and rescued effects of miR-3619-5p
Thereafter, the authors focused on the target gene of miR-3619-5p. Screened by CLIP-Data ≥5, Degradome-Data ≥2, and Program Number ≥3, 13 candidates were the probable targets of miR-3619-5p. However, only ARL2 expression was reduced by miR-3619-5p overexpression in both MHCC97 and Huh7-H cells (Fig. 3A and Supplementary Fig. S2F). In addition, GAS6-AS2 knockdown caused a decrease in ARL2 expression at both mRNA and protein levels (Fig. 3B and Supplementary Fig. S3A). Also, ARL2 was highly expressed in Huh7 and MHCC97 cells and further upregulated in these two HCC cells after heating treatment (Fig. 3C and Supplementary Fig. S3B). Significantly, RIP assay unveiled that GAS6-AS2, miR-3619-5p, and ARL2 were all strongly enriched by anti-Ago2 rather than by anti-IgG (Fig. 3D). Moreover, the putative binding sites between ARL2 and miR-3619-5p as well as the mutated ARL2 sequences were exhibited (Fig. 3E). It was revealed by the RNA pull down assay that ARL2 was remarkably pulled down by Bio-miR-3619-5p-WT but not by Bio-NC or Bio-miR-3619-5p-Mut (Fig. 3F). The follow-up luciferase reporter assay verified that miR-3619-5p bound with ARL2 at the predicted sites (Fig. 3G). More importantly, the authors discerned that inhibition of miR-3619-5p restored the suppressive effects of silenced GAS6-AS2 on ARL2 expression (Fig. 3H, I and Supplementary Fig. S3C). Afterward, they enhanced ARL2 expression in MHCC97-H and Huh7-H cells via pcDNA3.1/ARL2 (Fig. 3J and Supplementary Fig. S3D), and then conducted a collection of rescue assays. It was disclosed that upregulation of ARL2 evidently restored miR-3619-5p overexpression-mediated suppressive effects on cell proliferation, migration, invasion, EMT, and stemness (Fig. 3K–M and Supplementary Fig. S4A–C). Thus, the authors concluded that ARL2 was the downstream mediator in miR-3619-5p-regulated cell biological functions.
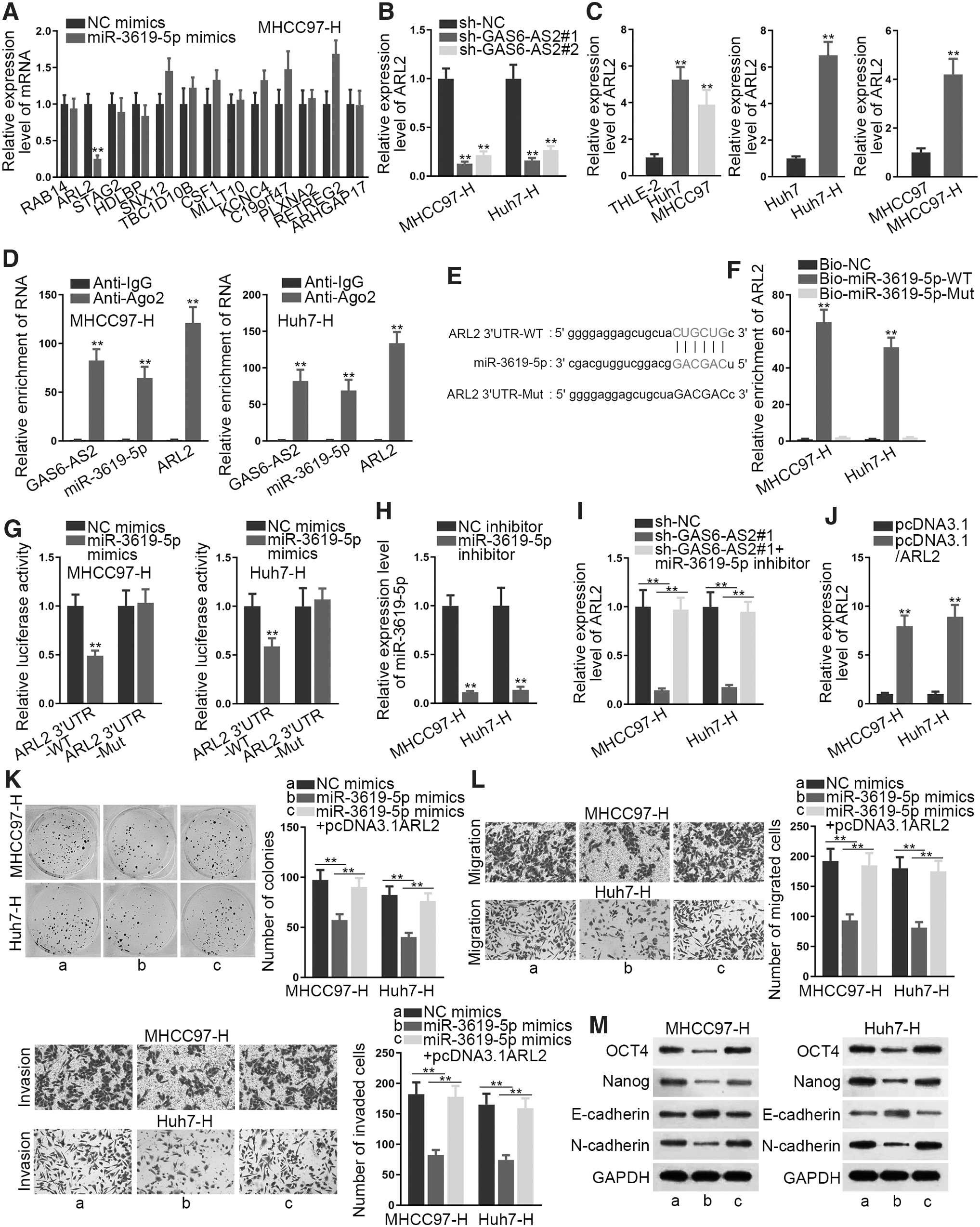
ARL2 was targeted by miR-3619-5p and rescued the effects of miR-3619-5p.
ARL2 was required in GAS6-AS2-mediated cell biological functions
The final rescue assays were implemented to explore whether ARL2 was acquired in GAS6-AS2-mediated effects in Huh7-H and MHCC97-H cells. As expected, ARL2 upregulation counteracted the restraining effects of silenced GAS6-AS2 on cell proliferation (Fig. 4A). Likewise, the abrogated migration and invasion in GAS6-AS2-silenced cells were restored by upregulated ARL2 (Fig. 4B). Also, GAS6-AS2 depletion-reduced cell EMT and stemness were offset by ARL2 overexpression (Fig. 4C, D, and Supplementary Fig. S4D, E). According to these data, ARL2 was the responsible mediator in GAS6-AS2-affected cell proliferation, migration, invasion, EMT, and stemness.
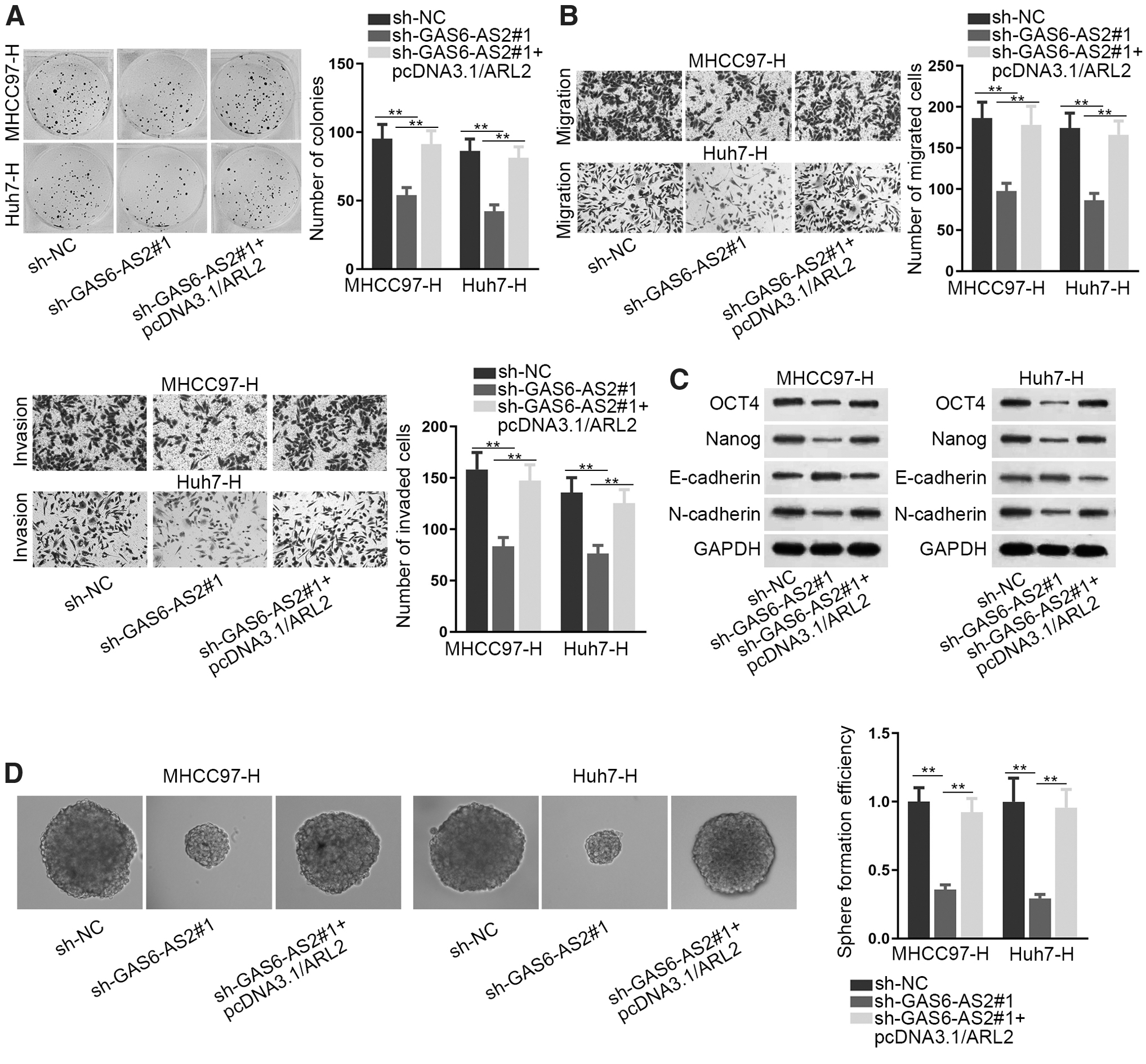
ARL2 was required in GAS6-AS2-mediated cell biological functions.
Discussion
RFA is currently the main local treatment for advanced unresectable HCC. However, insufficient RFA accelerates HCC cell invasion and migration. Present study examined the role of GAS6-AS2 in HCC after insufficient RFA, which has never been explored before. After they have established the Huh7-H and MHCC97-H cells via water bathing, they found out that GAS6-AS2 was upregulated in HCC cells after heating. Also, GAS6-AS2 promoted cell proliferation, migration, invasion, EMT, and stemness in Huh7-H and MHCC97-H cells. Since the cytoplasmic GAS6-AS2 was abundantly enriched in the RISCs, the potential ceRNA role of GAS6-AS2 was uncovered. Then, miR-3619-5p was identified to be sponged by GAS6-AS2. MiR-3619-5p was downregulated and inhibited cell proliferation, migration, invasion, EMT, and stemness characteristic in heated HCC cells. Previously, miR-3619-5p was verified as a tumor-inhibitor in multiple cancers. Tan A has pointed out that circZFR elevates CTNNB1 expression by sponging miR-3619-5p to trigger Wnt/β-catenin pathway and further accelerates HCC progression. 13 Zhang et al. has revealed that miR-3619-5p targets KPNA4 to suppress cell proliferation and elevate cisplatin sensitivity in cutaneous squamous cell carcinoma. 14 MiR-3619-5p directly targets β-catenin and CDK2 to activate p21, thus hampering bladder carcinoma development. 15 MiR-3619-5p activates CDKN1A expression to hinder cell growth in prostate cancer. 16 ARL2 has been widely reported as the tumor facilitator in various cancers. Long et al. have presented that miR-214 binds ARL2 to suppress colon cancer. 17 MiR-214 works as an antioncogene to suppress cell proliferation, migration, and invasion in cervical cancer through interaction with ARL2. 18 Here, the authors identified ARL2 as the direct target of miR-3619-5p. MiR-3619-5p negatively regulated ARL2 expression, while GAS6-AS2 elevated ARL2 expression via serving as the endogenous sponge of miR-3619-5p. ARL2 upregulation completely rescued the effects of miR-3619-5p upregulation or GAS6-AS2 depletion on the malignant behaviors of heated HCC cells. In a word, present study originally unveiled that GAS6-AS2 worked as a ceRNA of ARL2 by sequestering miR-3619-5p in in vitro model of HCC after insufficient RFA.
Apart from the study, miR-3619-5p or ARL2 associated ceRNA mechanism is also reported in other studies. LncRNA LINC00342 sponges miR-3619-5p to antagonize its inhibition on HDGF, further promoting infantile hemangioma growth. 19 It has been pointed out by Li et al. that ARL2 is involved in lncRNA ANRIL-mediated mitochondrial function of HCC via the ceRNA pattern. 20 Also, UCA1 enhanced mitochondrial function and cell viability in bladder cancer through upregulating ARL2 via sponging miR-195. 21 LncRNA LINC00202 serves as the endogenous sponge of miR-3619-5p to elevate RIN1 expression so as to promote retinoblastoma tumor progression. 22 The positive feedback loop of LINC01410/miR-3619-5p/FOXM1 axis facilitates cell proliferation and hampers apoptosis in papillary thyroid carcinoma. 23
On the whole, present study unveiled a novel ceRNA mechanism of GAS6-AS2/miR-3619-5p/ARL2 axis in aggravating in vitro HCC after insufficient RFA, which might help the development of treatment for patients with such disease.
Conclusions
The study unveiled a novel ceRNA mechanism of GAS6-AS2/miR-3619-5p/ARL2 axis in aggravating in vitro HCC after insufficient RFA, which might help the development of treatment for patients with such disease.
Footnotes
Acknowledgments
The authors sincerely appreciate all the laboratory members.
Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.