
Abstract
α-Emitting radionuclides have been approved for cancer treatment since 2013, with increasing degrees of success. Despite this clinical utility, little is known regarding the mechanisms of action of α particles in this setting, and accurate assessments of the dosimetry underpinning their effectiveness are lacking. However, targeted alpha therapy (TAT) is gaining more attention as new targets, synthetic chemistry approaches, and α particle emitters are identified, constructed, developed, and realized. From a radiobiological perspective, α particles are more effective at killing cells compared to low linear energy transfer radiation. Also, from these direct effects, it is now evident from preclinical and clinical data that α emitters are capable of both producing effects in nonirradiated bystander cells and stimulating the immune system, extending the biological effects of TAT beyond the range of α particles. The short range of α particles makes them a potent tool to irradiate single-cell lesions or treat solid tumors by minimizing unwanted irradiation of normal tissue surrounding the cancer cells, assuming a high specificity of the radiopharmaceutical and good stability of its chemical bonds. Clinical approval of 223RaCl2 in 2013 was a major milestone in the widespread application of TAT as a safe and effective strategy for cancer treatment. In addition, 225Ac-prostate specific membrane antigen treatment benefit in metastatic castrate-resistant prostate cancer patients, refractory to standard therapies, is another game-changing piece in the short history of TAT clinical application. Clinical applications of TAT are growing with different radionuclides and combination therapies, and in different clinical settings. Despite the remarkable advances in TAT dosimetry and imaging, it has not yet been used to its full potential. Labeled 227Th and 225Ac appear to be promising candidates and could represent the next generation of agents able to extend patient survival in several clinical scenarios.
Introduction
Targeted alpha therapy (TAT) is an attractive therapeutic option in patients with multiple micrometastases. It offers many advantages, including easy administration, the ability to treat multiple lesions simultaneously, and the possibility of combination with other therapeutic approaches for improved efficacy. 1 TAT utilizes an α particle-emitting radionuclide, primarily for treatment of cancer but also, much less commonly, in some other diseases, such as HIV, bacterial, and fungal infections. 2,3
TAT is typically delivered by attaching an α-emitting radionuclide to a biological molecule with targeting capabilities, such as a monoclonal antibody (mAb), with the help of a bi-specific chelating agent. This selectively delivers a high radiation dose directly to the target, with generally limited toxicity to the surrounding normal tissues. 4 Advances in the understanding of tumor biology, together with progress in mAb technology, chemical labeling techniques, and other related disciplines, are providing significant advances in the development of new clinical applications of α-emitting radionuclides in novel therapeutic agents.
The concept of clinical targeted therapy is not new and has been discussed since the “magic bullet” concept was first presented by Paul Erlich at the beginning of the 20th century. 5 As a key example, hyperthyroidism and thyroid cancer have been successfully treated using 131I for over 80 years. 6 Other β−-emitting radionuclides, including samarium-153 (153Sm) and stromtium-89 ( 89 Sr), have been used for palliative treatment of bone metastases since the middle of 20th century. 1 Remarkably, radium-223 (223Ra) is the first radiopharmaceutical agent to demonstrate improved overall survival among castration-resistant prostate cancer patients with symptomatic bone metastases, with mild side effects owing to its localized dose deposition. 7 Its clinical use was approved by the Food and Drug Administration (FDA) in 2013, as Xofigo® (223RaCl2, formerly Alpharadin) from Bayer Healthcare after the publication of the ALSYMPCA trial. Recently, a new class of therapeutic radiopharmaceuticals for TAT has been proposed. They consist of an α-emitting radionuclide (225Ac or 227Th) complexed to a chelator conjugated to an mAb.
Despite the expanding interested in TAT, in many preclinical and clinical studies, dosimetry and biological information at the microscopic level are sparse or not accurately defined. 8 A better understanding of these factors will provide better tools to maximize the benefits of TAT, avoid undesired toxicity, and determine the optimal quantity of an α-emitting radiopharmaceutical to be injected to a given patient. Similarly, routine clinical use of TAT will require new approaches with respect to dosimetry and imaging to monitor treatment efficacy.
TAT therapy has been shown to be a potent weapon in the treatment of some cancers, and many novel refinements are now being published, expanding the number of potential applications. Therefore, it is timely to provide an overview of recent progress in the field of TAT research.
Radiobiological Properties and Dosimetry of α-Therapy
An α particle is an energetic (4–9 MeV) positively charged helium-4 ( 4 He) nucleus, with a +2 electrical charge. 9 They have a short path length, typically <100 μm, which is characterized by a high linear energy transfer (LET) (60–230 keV/μm), depositing 1500 times more energy per unit path length than a β− particle. 10 Simulations and experiments have shown that a few α particles crossing a cell nucleus are enough to kill a cell, whereas more than 10,000 β− particles are needed to achieve the same biological effect. 11 This combination of short range and highly cytotoxic interactions means α particles primarily kill target cells by self-irradiation, offering true selectivity with only minor collateral damage to nearby healthy tissues. 12
This biological effectiveness of high LET radiation, in particular α particles, is understood in terms of their propensity to induce complex DNA double-strand breaks (DSBs) and multiple clusters of DSBs in target cells, making cellular repair mechanisms ineffective. 13 Also, the greater efficacy of α particles has been demonstrated to be independent of the oxygenation status of the cell. Conversely, low LET radiotherapy is less effective in hypoxic cells and at low dose rates. Values for relative biological effectiveness (RBE) for in vitro and in vivo cell survival have been reported ranging from 3 to 8 for α particles, indicating that, for an equal amount of energy deposited in a cell, the probability of cellular death is much greater than for low LET radiation. 14 Radiobiological characterization of targeted radionuclide therapy has been reviewed by Pouget et al. 8,15
However, the relevant radiobiology of α particle radionuclides is far from being entirely understood and new insights are needed to maximize the effectiveness of this new modality.
Moreover, given the complex and stochastic nature of α particle interactions, their dosimetry presents a number of challenges. Although most of α-emitting radionuclides can be imaged in γ cameras, at present, neither treatment planning nor post-therapeutic dosimetry are performed in clinical practice, due to the technical limitations of these techniques. However, it has been an active area of research with several advances in clinical imaging and dosimetry in recent years. 16
As stated above, following the in vivo biodistribution and pharmacokinetics of 223Ra is feasible by γ camera imaging, but poor spatial resolution makes it impossible to produce accurate quantitative information at a cellular scale. 16 Nevertheless, this can still produce useful clinical information. Hindorf et al. defined the imaging characteristics of 223Ra and its feasibility, 17 which enable the study of biodistribution and pharmacokinetics in human patients. 18,19 Also, Abou et al. confirmed in animal models that 223Ra is deposited at the bone surface surrounding the tumor and skeletal accumulation was dependent on the local blood vessel density. 20 A correlation has been demonstrated between uptake of 223RaCl2 with that of 99mTc and 18 F, which was also associated with the absorbed dose in the lesion. 21,22 Recently, studies have also been conducted to explore the usefulness of SPECT/CT 223Ra. 23,24
With regard to dosimetry, most conventional approaches are limited to average parameters such as absorbed dose to a macroscopic volume. Current dosimetry calculations predict a much higher level of hematotoxicity than experienced clinically. For example, a phase I clinical trial where patients received up to 925 kBq of 213Bi reported α particle doses of 1 Gy to the blood system. However, from microdosimetric calculations, the actual dose to important cell targets was 2 cGy for endothelial cells and 10 cGy for lymphocytes, <10% of the macroscopic dose to the blood. 9 This highlights that conventional dosimetric approaches are not always valid, highlighting the need to understand the distribution of dose at the cellular level to accurately predict biological effects. 25 Meaningful dosimetry studies with TAT require detailed information on the geometry of the target, as well as pharmacokinetic data of the α-emitting radionuclide and possible fate of daughters at a cellular and subcellular scale. 14 Usually, this is achieved with geometrical approximations and mathematical modeling to describe the uptake, clearance, and concentration of the radiopharmaceutical.
Palm et al. constructed a model to optimize radionuclide therapy, using radionuclide pharmacokinetics to estimate the absorbed dose in different tissues. 26 Hobbs et al. developed a bone marrow toxicity model for 223RaCl2 and concluded that cellular scale dosimetry is necessary to explain the good toxicity profile of 223Ra clinically observed. 27 Recently, Moreira et al. modeled different tumor growth models and three possible 223Ra uptake scenarios to determine the most realistic scenario based on clinical data from the ALSYMPCA trial. 28 The authors concluded that only a subpopulation of target cells is directly affected by 223Ra with strong evidence of saturation in these hot spots. These data confirm that TAT has not yet been used to its full potential, and using body weight to calculate the administered activity in a treatment fraction may potentially lead to many patients being overtreated (high toxicity) or undertreated (no clinical effect). Furthermore, Taprogge et al. showed by mathematical models that retention of 223Ra in the human skeleton requires two different compartments, with an initial location to the bone surface followed by incorporation into the bone matrix. 29
Off-Target Effects of α Particles
It is clear from the physical characteristics of TAT that due the short path length of α particles relative to target dimensions and incomplete targeting, some cell nuclei receive multiple hits, while others receive no direct irradiation, giving rise to an inhomogeneous irradiation profile. 30 Those cells that are not directly irradiated may diminish the overall therapeutic benefit. Because of this, early studies had predicted some advantage of β−-emitting radionuclides in macrolesions due to the cross-fire effect (dose delivered outside the targeted cell due to long particle ranges), reducing problems associated with heterogeneous uptake in the target. 31
However, biological off-target effects have been observed in response to ionizing radiation, including high LET irradiation. These are frequently termed “the bystander effect” and are most commonly seen as cell killing occurring in cells that see no direct irradiation. 32 These effects may particularly important in heterogeneous regimens, where indirect killing of nontargeted cells is required to ensure maximal therapeutic benefit. In addition to local bystander effects, longer-range adaptive effects, mostly inflammatory responses in distant tissues, have also been reported. In some preclinical studies, bystander effects have been as effective as the direct effect after low-dose exposure, suggesting that under these conditions and in a low-dose rate scenario, a bystander response could have a large impact on overall effectiveness. 33 These effects are not currently considered in conventional treatment planning.
A series of elegant studies has clarified the role of bystander effects after irradiation with several radionuclides. In 2002, Xue et al. showed contradictory bystander responses in the same in vivo model after treatment with 125I and 123 I, short-range Auger emitters with substantial differences in half-life (13.3 h and 60.5 d, respectively). 34
In a different study, Boyd et al. compared the bystander effect induced by different radiation qualities. 35 In particular, they compared the effectiveness of indirect effects following external beam γ radiation exposure with that following exposure to a β− emitter (131I), an Auger emitter ( 123 I), or an α emitter (211At) in two different tumor lines. The standard bystander response mediated by external beam irradiation is an increase at low doses followed by a plateau. In contrast, treatment of cells with an α-emitting radionuclide led to a U-shaped response curve, with an increased cell kill in the recipient cells at low activities, but a decreased effect at increasing activities. This U-shape bystander curve response was also reported by Karthik et al. in human lymphocytes exposed to external α particles. 36 Finally, Ladjohounlou et al. reported that bystander effects contributed up to 36% of cell killing when α emitters were used in in vivo and in vitro experiments 37
Understanding the mechanism of bystander effects may help TAT achieve its full potential and have profound implications on the clinical applications of radionuclides. Bystander effects in in vivo and in vitro radionuclide therapy have been reviewed by Brady et al. 38 In addition, there is now evidence that TAT can stimulate the immune system to deliver a better and more robust antitumor response. 39,40
Another mechanism by which α-emitting radionuclides may overcome heterogeneous uptake is through tumor antivascular α therapy (TAVAT) as shown by Allen et al. 41 TAVAT assumes that α-emitting radionuclides deposit energy in multiple hot spots near capillaries in the target volume, causing closure of capillaries and subsequent reduction of oxygen and nutrients, leading to regression in solid tumors.
New TAT Radionuclides
The approval of 223RaCl2 for clinical use has stimulated clinical interest in TAT, with an increasing number of clinical trials around the world. α-Emitting radionuclides, which have been proposed for therapeutic application and are undergoing clinical investigation include Astatine-211 (211At), Bismuth-212 (212Bi), Bismuth-213 (213Bi), Actinium-225 (225Ac), and Thorium-227 (227Th), summarized in Table 1. Production of these radionuclides ranges from nuclear reactors to cyclotrons and generator systems. There is a continuous effort to develop simpler and more efficient production methods to make α-emitting radionuclides more widely available. Another key aspect of TAT is the need to establish efficient chemical procedures to bind the α radionuclide to a suitable carrier. The merits of these different α emitters are summarized below.
Main Characteristics of the Potential α-Emitting Radionuclides for Clinical Applications
211At, Astatine-211; 212Bi, Bismuth-212; 213Bi, Bismuth-213; 225Ac, Actinium-225; 227Th, Thorium-227.
Astatine-211
Astatine-211 is a radiohalogen with a half-life of 7.2 h and offers many potential advantages for TAT due to its average range in soft tissues of 57 μm, average LET of 97 keV/μm, and single α particle emission per decay. However, its use is constrained by its limited current availability. 42 211At decays through two different paths, through electron capture (58%) with emission of 77–92 keV X-rays or through emission of an α particle (42%) with energy of 5.87 MeV (Fig. 1). The decay product of the electron capture, Polonium-211 (211Po T 1/2 = 0.52 s), then decays by α emission (100%) with energy of 7.45 MeV and a range in soft tissues of 71 μm. 43 Therefore, each 211At decay results in one α particle with mean energy of 6.78 MeV, leaving stable 207Pb. 43

Decay scheme of Astatine-211. The T 1/2 is shown inside of each box, and between boxes, the energy of α transition (MeV) and the percentage of the branch when necessary. The chemistry of astatine allows radiolabeling with a molecule of interest. Solid arrows represent α decay for the nuclear transition. Dashed arrows represent any other type of decay, mostly β− emission or EC for the nuclear transition.
Although 211At can be produced by α bombardment of enriched bismuth through the 209Bi(α,2n)211At nuclear reaction, the availability of cyclotrons offering α particle beams of sufficient energy (20–30 MeV) is limited. Typically, isolation of 211At from bismuth is performed either by dry distillation or by wet chemical approaches. Currently, 211At is routinely produced through cyclotron nuclear reaction in the United States (Seattle, Bethesda, and Durham) and Denmark (Copenhagen). Even with his short half-life, it can be distributed to centers distant from production sites.
Alternative methods for producing 211At involve production of Radon-211 (211Rn), which can be obtained by proton spallation of uranium or thorium targets or by irradiation of natural bismuth with lithium-7 ( 7 Li) atoms. 211Rn/211At generators may facilitate easier distribution due to the longer half-life of 211Rn (T 1/2 = 14.6 h). 44
The inherent cost for production of 211At is relatively inexpensive and comparable to that of commercially available β emitters.
The 7.2h half-life of 211At is acceptable for chemical labeling and clinical pharmacokinetics even with slower target tissue uptake. Another advantage is that its second decay branch to 211Po emits X-rays with energy of 77–92 keV, enabling biodistribution and dosimetry studies. Currently, its radiolabeling chemistry with proteins and antibodies is the subject of extensive studies in preclinical trials. Nevertheless, in vivo instability of the labeled compound and scarce availability of the radionuclide remain major challenges with 211At. 42
Elgqvist et al. investigated the therapeutic efficacy of 211At labeled with a fragment of an mAb in a preclinical model of ovarian cancer. 45,46 The results of these studies showed that dosimetry to the cell nuclei is strongly dependent on the tumor size—while in small tumors (radius <30 μm), all cells are eradicated, in tumors with radii >75 μm, there is a small fraction of unirradiated cells in the core that explain the low tumor-free fraction, despite the high mean absorbed dose in the tumor [>22 Gy for an injected activity of 400 kBq of 211A-MX35 F(ab′)2]. 46 Previously, Charlton reported similar results using Monte Carlo simulation. 47
Moreover, promising results were also obtained in two Phase I trials of 211At conjugated for treatment of malignant gliomas or ovarian cancer, which confirmed its safety and failed to identify a dose-limiting toxicity. 48,49 Several radiolabeled complex antibodies with 211At have been shown to be selective in different preclinical models, and highly cytotoxic for several types of cancer. 50 –54
Recently, a novel α-emitting radiopharmaceutical was developed by incorporating 211At into the chemical structure of a small-molecule PARPi (211A-MM4). This novel radiopharmaceutical showed promising efficacy both in vitro and in vivo. 55 In addition, it was shown that low-dose-fractionated therapy improved its toxicity profile in animal models and gave a more durable antitumor response. Different cell lines also demonstrated differential sensitivity to α particle irradiation in vitro, suggesting sensitivity to this agent depended on underlying biology. Fractionated treatment with 211At labeled to the anti-prostate stem cell antigen A11 minibody also showed strong tumor growth inhibition on both macrolesions and microlesions. 52 There is also an ongoing phase I/II clinical study using 211At labeled to anti-CD45 antibody for the treatment of acute myeloid leukemia (AML) and acute lymphoblastic leukemia, before bone stem cell transplant. 42
Bismuth-212 and Bismuth-213
Bismuth-212 with a half-life of 60.6 min is a hybrid α/β emitter with an α emission with mean energy of 7.8 MeV available from the decay of 228Th to stable 208Pb. 56 Since its half-life is too short for realistic transportation between sites, a generator that uses 224Ra as parent radionuclide provides on-site production of 212Bi for radiolabeling of target vectors. As its half-life is very short, making radiopharmaceutical preparation challenging, the idea of an in vivo 212Pb/212Bi generator came, as 212Pb is a longer-lived parent (T 1/2 = 10.6 h) and decays into 212Bi after emission of an electron. 57 (Fig. 2A). The 10.6h half-life of 212Pb allows time for preparation of the radiopharmaceutical as well as tissue targeting before much of the 212Bi is produced and decays. 212Pb has demonstrated significant utility in both in vitro and in vivo models. 57 The major disadvantage of 212Bi is the high-energy gamma (γ) emission of 208Tl (E γ = 2.6 MeV), which requires heavy shielding to minimize radiation exposure, thereby limiting the clinical utility of this radionuclide.
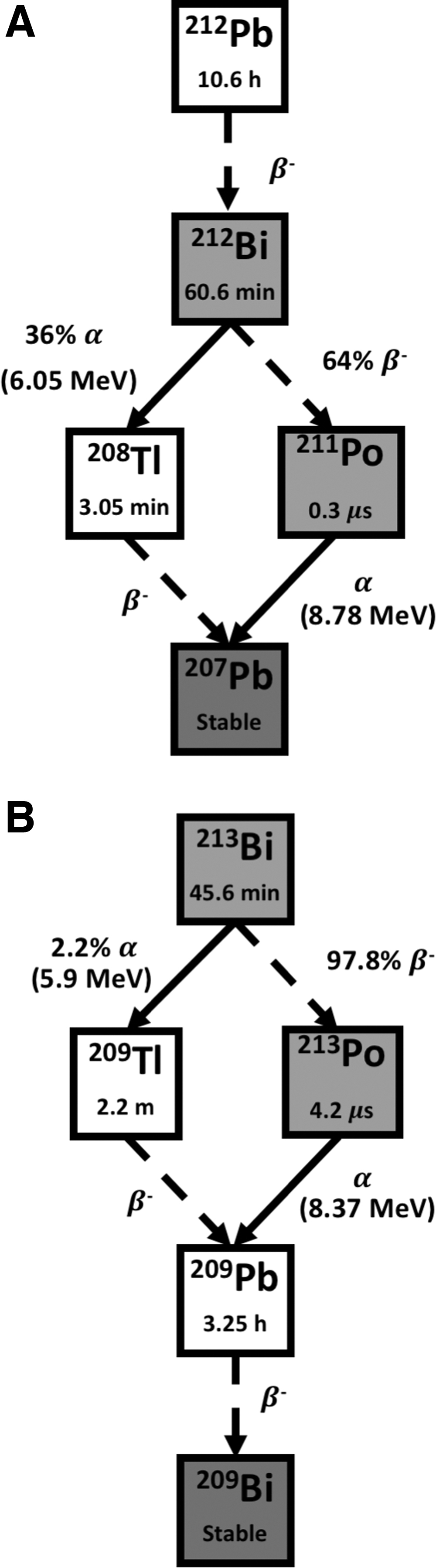
There are some studies of 212Bi radiolabeled with tumor-specific antibodies reporting prolonged survival compared with controls. 58,59 So far, no clinical trials have been reported for 212Bi compounds. However, a phase I clinical trial, which investigated the use of 212Pb-trastuzumab for treatment of human epidermal growth factor receptor 2 positive (HER2+) ovarian, colon, and pancreatic tumors, has been completed, showing minimal toxicity. 60
Bismuth-213 is also a hybrid α/β− emitter with a half-life of 45.6 min, derived from the α-decay of 225Ac. 56 The major disadvantage is the cost, as large amounts are needed to attain an effective treatment due to the short half-life. 61 213Bi predominantly decays through β− emission to the ultra-short lived 213Po (T 1/2 = 4.2 μs), which in turn decays by pure α emission with energy of 8.37 MeV. The remaining 2.2% decays to Thallium-209 (209Tl) through α particle emission (E α = 5.55 MeV, 0.16%, and E α = 5.9 MeV, 2.01%). 62 Finally, 209Tl decays through β− emitter 209Pb (T 1/2 = 3.25 h) into stable 209Bi (Fig. 2B). Most of the total particle energy emitted in each 213Bi disintegration originates from α decay (92.7%), which drives its cytotoxic effects, while only 7.3% of decay energy is contributed by β− emission. In addition, the decay of 213Bi is accompanied by a 440 keV photon emission (probability of 26.1%) that allows 213Bi biodistribution to be monitored for dosimetry and pharmacokinetic studies. The major impediment for its clinical use has been concern regarding the natural accumulation of free bismuth in the kidneys. 63
The therapeutic potential of 213Bi labeled with antibodies and peptides has been demonstrated in patients with leukemia. 59 In addition, 213Bi labeled with plasminogen activator inhibitor type 2 (PAI2) has been reported to inhibit tumor growth in prostate and breast cancer models, resulting in a dose-dependent tumor growth inhibition with minimal toxicity up to 3.7 MBq of systemically administered 213Bi-PAI2 in mouse models. 64,65 Derrien et al. showed a survival benefit of labeled 213Bi in mouse models of peritoneal carcinomatosis of ovarian origin. 66 A phase I trial for AML treatment showed feasibility and safety with activities up to 37 MBq/kg being well tolerated. 67 A subsequent Phase I/II trail with sequential administration of chemotherapy (cytarabine) and 213Bi-lintuzumab has concluded it is well tolerated and capable of inducing remission in patients with AML. 68
In addition, TAT with 213Bi-labeled DOTATOC in 25 patients with challenging neuroendocrine tumors refractory to therapy with β− emitters resulted in a long-lasting antitumor response. 69 Thus, in a similar concept 213Bi prostate-specific membrane antigen (PSMA)-617 was administrated in two cycles in a patient with metastatic castrate-resistant prostate cancer (mCRCP), which was progressive following conventional therapy. With a total delivered activity of 592 MBq, these patients also demonstrated biochemical response and remarkable tumor regression. 70 Furthermore, 213Bi-labeled substance P analog has been tested by locoregional administration in 67 patients with grade II–IV glioma. 71 –73 Treatment was fractionated in up to eight cycles at 2-month intervals, with remarkable tumor regression.
Recently, a pilot study used intravesical administration of 213Bi-labeled anti-EGFR in 12 bladder cancer patients who were resistant to standard therapy, and showed remarkable tumor remission with a safe toxicity profile. 74
Radium-223
Most current research activity in TAT is focused on 223Ra. As a calcium mimetic, 223Ra has an intrinsic affinity to bone, forming complexes with the bone hydroxyapatite crystals at sites of high bone turnover, with almost no redistribution of daughter nuclides. 75
223Ra has a physical half-life of 11.4 days with a six-stage decay chain, emitting four α particles with mean energy of 5.87 MeV and tissue penetration of 54 μm before reaching the stable 207Pb. 223Ra releases 95.3% of its energy as α particles and 3.6% as β− particles with very little (1.1%) γ emission (Fig. 3). 7 223Ra is generally artificially produced, through a generator from the 227Ac (T 1/2 = 21.8 years) parent. 76
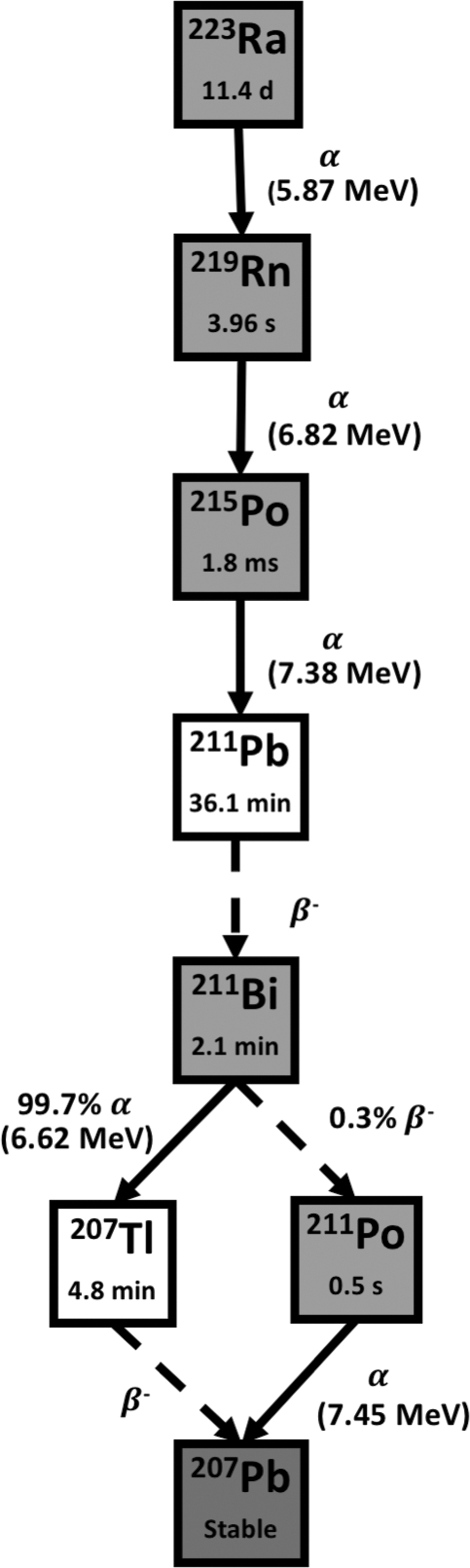
Decay scheme of Radium-223. The T 1/2 is shown inside of each box, and between boxes, the energy of α transition (MeV) and the percentage of branch when necessary. Due to its poor chemistry, 223Ra has not yet been successfully combined with targeting molecules. Solid arrows represent α decay for the nuclear transition. Dashed arrows represent any other type of decay, mostly beta emission or EC for the nuclear transition.
The long physical half-life of 223Ra provides appropriate time for its preparation, long distance distribution, and administration to patients. Its low γ emission is favorable from the point of view of handling, radiation protection, and treatment on an outpatient basis. Nonetheless, due to its poor chemistry, the availability of appropriate chelating agents with sufficient in vivo stability is an issue for applications other than treatment of bone metastases. The maintenance and biological follow-up of the decay daughter products continue to be a continuing challenge. 61
A major milestone was reached in the ALSYMPCA study, which demonstrated a survival benefit of 223Ra admission together with a favorable safety profile. 7 223RaCl2 (Xofigo) was approved for clinical use by the FDA and European Medicines Agency in 2013 and has been widely used for men with symptomatic mCRPC. Its peak skeletal uptake occurs within 1 h of injection, with no subsequent redistribution. Its blood clearance is rapid after intravenous administration and the skeletal uptake is estimated to range between 40% and 60% of the administered activity. 77 In addition, non-negligible levels of 223RaCl2 can be found in the spleen, stomach, and intestinal organs that rapidly transit to the gastrointestinal tract for excretion. 78
In clinical reports 223RaCl2 had a pain response in up to 71% of patients, and had significant positive effects in bone alkaline phosphatase and prostate-specific antigen levels and overall survival. 79,80 Data from these studies showed that there was no evidence of long-term toxicity during the 2-year follow-up and the most common forms of adverse events were transient diarrhea, fatigue, nausea, and vomiting. 75
The improvement in overall survival and low toxicity of 223Ra open the possibility for it to be combined with other modalities, such as docetaxel or external beam radiotherapy, and the extension of its application to skeletal metastases from other primary tumors. However, questions remain regarding its treatment schedule and dosing for optimized patient benefit, as the dosing and scheduling used in the ALSYMPCA trial were not necessarily optimal. 81
Thus, a new phase II trial was performed to compare standard regimen (55 kBq/kg every 4 weeks for 6 cycles) against higher activity (88 kBq/kg every 4 weeks for 6 cycles) or for an extended schedule (55 kBq/kg every 4 weeks for 12 cycles). However neither, of these alternative regimens was more effective than the standard 223RaCl2 schedule. 81
Suominen et al. provided insights into the mechanisms of action of 223RaCl2 showing a dose-dependent inhibition of osteoclast differentiation, data that support the dual effect of 223RaCl2 on the cancer cycle by targeting both osteoclast and tumor cells. 82
Malamas et al. showed 223RaCl2 treatment stimulated the immune system by inducing T cell-mediated cell death in different cancers. 39 Subsequently, a phase II trial of patients with advanced breast cancer and progressive bone-dominant disease demonstrated that 223Ra has selective uptake in areas of increased bone metabolism in these patients, and one-third of target lesions showed significant metabolic decrease. 83 A phase I/II clinical trial testing the feasibility and safety of the antimetabolite chemotherapy agent Capecitabine plus 223Ra is ongoing in patients with advanced breast cancer. 84 Subbiah et al. also extended the application of 223RaCl2 to osteosarcoma with a phase I escalation trial finding minimal hematologic toxicity and recommending a follow-up phase II study with a monthly activity of 100 kBq/kg. 85
Despite the apparent additional benefits of 223RaCl2 plus hormonal therapy, the ERA-223 trial showed that 223RaCl2 should not be administered in combination with abiraterone plus prednisone or prednisolone as it significantly increases the risk of osteoporotic fractures. 86,87 It is hypothesized that increased fracture risk is the result of multifactorial insults to bone microenvironment from androgen deprivation, ionizing radiation, and prednisone. 88 However, concomitant use of bone-protecting agents dramatically decreased the risk of osteoporotic fractures in the ERA-223 trial, and are now required in future combination studies with 223Ra.
The ERA-223 trial highlighted the importance of controlled trials to assess the safety and benefit of combination therapy. Many 223RaCl2 clinical trials are currently active and recruiting (Supplementary Table S1). The main focus is on long-term follow-up, new scheduling, expansion of clinical settings, and combination of 223RaCl2 with other agents. These combination studies mainly focus on five combination strategies: abiraterone, enzalutamide, docetaxel, immunotherapy, and PARP inhibitors. 89
223RaCl2 in combination with paclitaxel was tested in a phase I trial of 15 patients with acceptable toxicity when combined with weekly administration of paclitaxel. 90 A similar phase I/II trial combining 223RaCl2 with docetaxel is ongoing. 91
In an elegant study, Leung et al. demonstrated the potential impact of bystander effects of 223RaCl2 therapy showing different cellular sensitivities in vivo. 92 However, in vitro studies have shown significant variation in cellular sensitivity across different tumor cell models after exposure to 223Ra, suggesting further work is needed before applying it to different cancers. 93,94
Actinium-225
Actinium-225 is a pure α emitter with a half-live of 10 days. It decays sequentially by α emission through three relatively short-lived daughter radionuclides, 221Fr (T 1/2 = 4.8 min), 217At (T 1/2 = 32.3 ms), and 213Bi (T 1/2 = 45.6 min), each of which then also emits an α particle with a large cumulative energy of 28 MeV. 95 Then 213Bi decays through β− disintegrations and α emission into stable 209Bi. Thus, the predominant decay chain of 225Ac yields four α particles, along with two β disintegrations of 1.6 and 0.6 MeV maximum energy (Fig. 4). 96 In addition, useful γ emission for in vivo imaging is generated in the 225Ac decay scheme from decay of 221Fr (218 keV, 11.6% emission probability) and 213Bi (440 keV, 26.1% emission probability). 95 Its long half-live of 10 days and the multiple α particles generated in the decay chain render 225Ac a highly cytotoxic radionuclide. 96
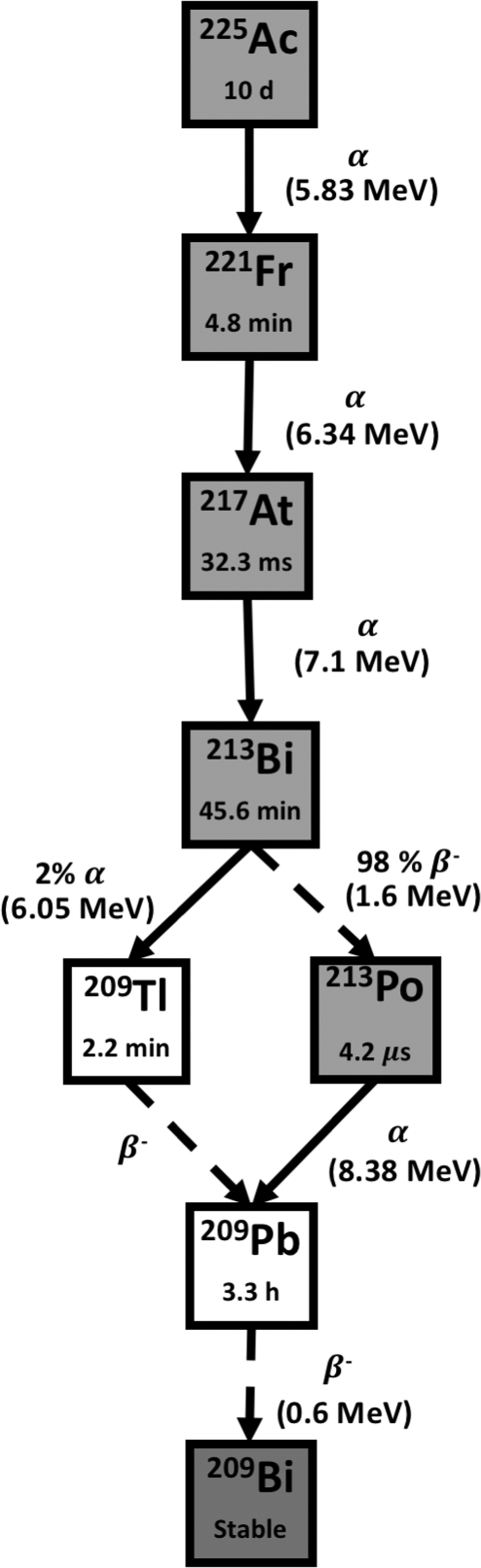
Decay scheme of Actinium-225. The T 1/2 is shown inside of each box, and between boxes, the energy of α transition (MeV) and the percentage of branch when necessary. The good chemistry of actinium allows radiolabeling with a molecule of interest. Solid arrows represent α decay for the nuclear transition. Dashed arrows represent any other type of decay, mostly beta emission or EC for the nuclear transition.
225Ac is mainly produced by isolation from 229Th, which is a decay product of 233U, yet the quantity available through this route is not sufficient for commercial applications as there is no large-scale production of 233U. 95 At present, viable sources of 229Th are available at the Directorate for Nuclear Safety and Security of the Joint Research Center, formerly known as Institute for Transuranium Elements (Germany), Oak Ridge National Laboratory (United States of America), and the Institute of Physics and Power Engineering (Russia). Overall, the average global production is 63 GBq of 229Th. 97 Alternatively, 225Ac can also be produced in cyclotrons delivering 18 MeV proton beams by bombardment of radioactive radium through the 226Ra(p,2n) reaction. 95
In theory, 225Ac could be as much as ∼100 times more potent than 213Bi-conjugated analogs for the same activity, by virtue of the α cascade to the target cell and the longer half-life of 225Ac, which is 313-fold greater than the 213Bi half-life. 98 An 225Ac decay on the nucleus deposits the same dose as four 213Bi decays. This increased potency provides a clinically relevant dose, even when activities as small as 37 kBq/kg of radionuclide are administrated. 99
However, the major impediment for its clinical use has been concern regarding the biological fate of its daughter radionuclides, particularly 213Bi, which accumulates in the kidneys. Issues relating to suitable chelating agents and the chelation chemistry to sequester this element in vivo remain to be solved, as none of the radiolabeling methods investigated so far has proven to efficiently form the required complex. 98
TAT using 225Ac conjugated to antibodies or other carriers are promising treatments for many malignant diseases such as myeloid leukemia, ovarian cancer, bladder cancer, melanoma, and non-Hodgkin's lymphoma. A successful phase I clinical trial with 225Ac linked to an anti-CD33 antibody demonstrated antitumor activity against AML and a safe toxicity profile at activities below 111 kBq/kg. 99 A phase I/II trial demonstrated the feasibility of fractionated-dose 225Ac-lintuzumab combined with the cytosine arabinoside chemotherapy agent cytarabine, inducing remission with activity of 74 kBq/kg per fraction. 100
After the encouraging results of 213Bi-DOTATOC, a follow-up investigation using 225Ac-DOTATOC was tested in 39 patients with progressive neuroendocrine tumors. This showed promising efficacy and the maximum tolerable activity was considered to be 40 MBq for a single cycle, or 25 MBq every 4 months and 18.5 MBq every 2 months for multiple fractions. 101
225Ac-PSMA-617 was studied in patients with mCRPC. 102 In Kratochwil et al., 40 heavily pretreated patients with advanced mCRPC were treated with three cycles of 100 kBq/kg of 225Ac-PSMA-617 with promising antitumor activity, supported by positive PSA decline (>50%) in 63% of patients, where severe xerostomia was the main side effect of treatment. 103 Despite the long physical half-life of 225Ac, this suggests the peptide targeting was sufficiently stable to provide the desired pharmacokinetic properties and prevent accumulation of free 225Ac in undesired organs. 104 Sathekge et al. extended the 225Ac-PSMA-617 treatment schedule to chemotherapy-naive patients, with good antitumor activity assessed by PSA level and imaging in 80% of patients, with mild and reversible xerostomia in all patients. These new data can potentially move TAT treatments into early-stage cancers. 105
In a comparative modeling study, 225Ac-PSMA produces doses to the target cells 900-fold greater than 177Lu-PSMA and 14 times more than 223RaCl2 per unit of activity retained in the target volume. 106 This study highlights the importance of subcellular localization of the α-emitting radionuclide as 225Ac and 223Ra have similar decay chains; however 225Ac-PSMA is found in the cytoplasm and membrane of cancer cells, while 223RaCl2 is located in the bone compartment surrounding the tumor.
A clinical trial in older patients with advanced myeloid leukemias treated with two activities of 74 kBq/kg 225Ac-labeled lintuzumab, anti-CD33 mAb, has demonstrated a 56% response rate with moderate side effects. 107 Another phase I/II trial is investigating treatment with 225Ac-labeled lintuzumab after chemotherapy to reduce leukemia burden in patients with AML (Supplementary Table S2).
Thorium-227
Thorium-227 has a physical half-live of 18.72 days, and is a α emitter with an average energy of 6.02 MeV, along with frequent γ emission (E γ = 236 keV, 11.2%, and E γ = 50 keV, 8.5%). 56 227Th decays through radioactive 223Ra and the other short-lived radionuclides in its decay chain to stable 207Pb, emitting a total of five α particles (Fig. 5). 227Th can be produced by natural decay of 227Ac (T 1/2 = 21.8 years). 227Ac can be produced in a reactor by neutron irradiation of 226Ra that produces 227Ra (T 1/2 = 42 min), which in turn decays by β− disintegration to provide the desired 227Th. The long half-life of 227Th allows for transportation and preparation of the radiopharmaceutical. 108 Like its daughter 223Ra, 227Th has also been of interest in the development of therapeutic bone-seeking radiopharmaceuticals. Furthermore, Algeta (Norway) [a subsidiary of Bayer (Germany), which developed Xofigo], is also developing 227Th-labelled antibodies for therapeutic use. Additional sources of 227Ac will be needed for worldwide availability of 227Th-labeled pharmaceuticals.
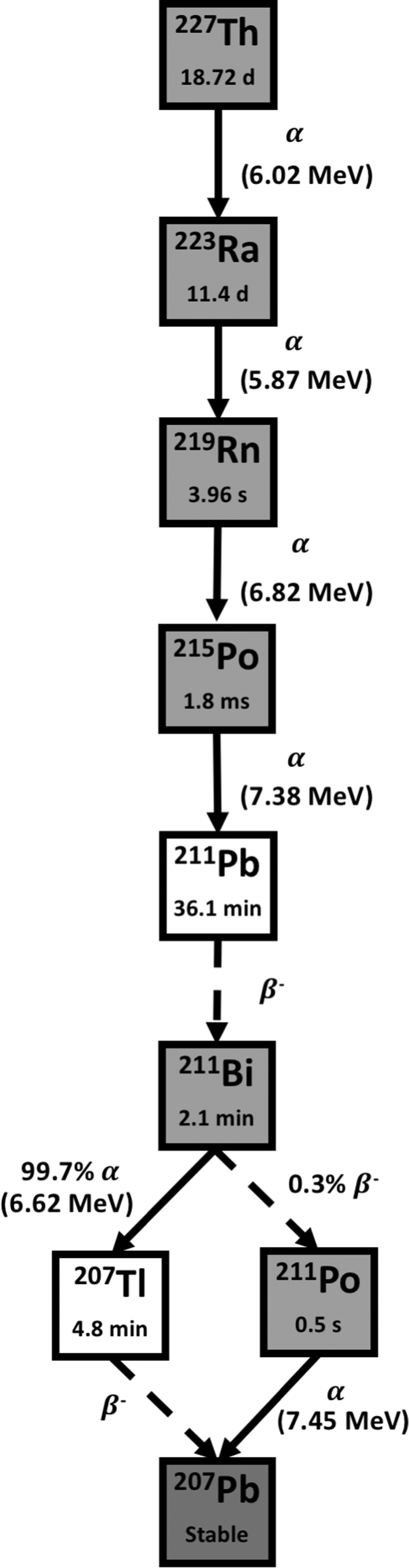
Decay scheme of Thorium-227. The T 1/2 is shown inside of each box, and between boxes, the energy of α transition (MeV) and the percentage of branch when necessary. The chemistry of Thorium allows radiolabeling with a molecule of interest. Solid arrows represent α decay for the nuclear transition. Dashed arrows represent any other type of decay, mostly β− emission or EC for the nuclear transition.
Due to its chemical properties, 227Th can be complexed to a variety of antibodies and proteins. Current research using 227Th conjugates centers on developing and testing new radiopharmaceuticals for the treatment of breast, lymphoma, renal, AML, ovarian, and prostate cancers (Supplementary Table S3). 40,109
Therapy using the anti-CD20 mAb rituximab labeled with 227Th was demonstrated to kill lymphoma cells in nude mice bearing human B-lymphoma xenografts, without any serous toxicity. 107 In addition, Dahle et al. demonstrated that 227Th-rituximab has a better therapeutic effect per unit of absorbed dose than 99 Y-tiuxetan-ibritumomab (Zevalin) or external beam irradiation. 110 The authors also suggested treatment fractionation through multiple injections can maximize the therapeutic effect per dose unit, while minimizing toxicity.
Preclinical studies of 227Th-trastuzumab for treatment of HER2+ positive ovarian and breast cancer showed encouraging antitumor effect without serious toxicity. 111,112 227Th-PSMA was shown to be selective and has a potent antitumor activity in vitro and in vivo. 113 These preclinical data encouraged the further investigation of this agent in an ongoing phase I trial in mCRPC.
Mesothelin (MSLN) targeting 227Th conjugate demonstrated a significant antitumor effect in vivo with good tolerability, showing reduced cellular viability with an upregulation of immune response. 114 These encouraging results led to a phase I clinical study on the use of 227Th-MSLN in patients with solid tumors known to express mesothelin. Furthermore, MSLN targeting 227Th conjugates have demonstrated synergistic antitumor effect when combined with DNA damage response inhibitors, particularly ATRi and PARPi in preclinical cancer models. 115
α Emitter Selection
The clinical use of 223RaCl2 showed clear benefits in overall survival in mCRPC, supporting the proposed theoretical therapeutic advantages of α particles over the more widely used β−. Other α-emitting radionuclides that are the subject of on-going research for therapeutic application include 211At, 212Bi, 213Bi, 225Ac, and 227Th.
The selection of an α-emitting radionuclide for radiopharmaceutical development requires careful consideration. It has been shown that it is possible to clinically use radionuclides with a broad range of physical half-lives, from 45 min to almost 19 days, as long as the physical half-life matches the pharmacokinetic parameters of the radiopharmaceutical and are appropriate for the desired application. As all α particles will completely lose their energy in <100 μm, corresponding to only a few cell diameters, the precise kinetic energy of the generated α particles is less critical for normal tissue sparing.
Other characteristics, in addition to the physical properties, should also be taken into consideration during the selection of the ideal radionuclide. These include both the feasibility and cost of radionuclide production, as well as the capability to easily manipulate the radionuclide to produce a radiopharmaceutical that is stable in vivo.
Half-life has a major impact in the logistics of production and distribution. An ideal radionuclide should permit widespread distribution from the production sites to all over the world. A longer half-life will ensure minimal waste due to decay during radiochemical processing and distribution of the radiopharmaceutical. However, radionuclides produced through a generator represent a convenient way to produce short-lived radionuclides at user sites through the decay of a long-lived parent, if suitable parents are available.
Radiopharmaceutical design remains a key challenge to optimizing therapeutic benefit. Many TATs are characterized by inhomogeneous distributions of the radiopharmaceutical, with some cells in the target potentially receiving no radiation. This implies that the radionuclide should be able to be produced with high specific activity for radiolabeling targeting molecules to ensure that sufficient dose is delivered to the target without saturating the limited number of receptors available at the target sites. 4 However, lower specific activity may be suitable for bone pain palliation and treatment of joint inflammation by radiation synovectomy.
Another important aspect that should be taken into account is the detachment of daughters from the targeting carrier due to the intense recoil energy upon α decay (>100 keV) that exceeds the energy of chemical bonds. The resulting free daughter nuclides can then diffuse from the target, leading to energy deposition in healthy tissue. 109 Palm et al. demonstrated, using microdosimetric calculations, that the energy deposited in the target could be reduced by 50% if diffusion of daughters of 211At was taken into account. 116 Furthermore, a study evaluating the anti-CD33 mAb lintuzumab 225Ac labeled for the treatment of leukemia in mice found that most of the radiation dose in the kidneys was due to renal uptake of free 213Bi released following α particle decay of 225Ac. 63 Therefore, it is of importance to study the fate of all radionuclides involved in the decay chain.
Despite the encouraging progress in TAT, treatments usually do not have optimal pharmacokinetics. Ideally, radiopharmaceuticals should have a rapid clearance from the blood stream with a robust tumor uptake. This led to the suggestion of a pretargeting approach, a concept proposed by Goodwin et al. in 1988. 117 This is a two-step procedure: in the first step, a pretargeting nonradioactive molecule is administrated and adequate time is given for its localization to the specific antigen in the target volume. Then, after clearance of the pretargeted molecule, the radiolabeled molecule is injected, and will rapidly diffuse in the target and specifically bind to the pretargeted molecule. This approach improves the target-to-normal tissue ratios of absorbed dose. The pretargeting approach is very promising for short half-life radionuclides such as 211At and 213Bi.
Development of a Novel α-Emitting Radiopharmaceutical
The development of novel radiopharmaceutical is a long and costly process, estimated to cost $800–1,700 million over 10 or more years. 118 The development process starts with target identification. After target identification, it is crucial to find the lead molecule to be radiolabeled. Following the lead molecule selection, biological properties of the radiopharmaceutical must be evaluated in vitro and in vivo, to evaluate affinity, saturability, stability, pharmacokinetics, and biodistribution. Further toxicology studies in animals are done to assure safety before translation into humans. Usually, phase I clinical trials include safety studies in healthy humans, although this is frequently unfeasible for radiopharmaceuticals. Phase II and III will define the settings and efficacy of the radiopharmaceutical. 119
In the particular case of development of novel radiopharmaceuticals for the treatment of mCRPC, molecular targets have been recently identified. PSMA is a transmembrane protein highly and specifically expressed at all tumor stages on the surface of cancer cells and its levels are correlated with the aggressiveness of the disease, making it an attractive target. 120
This antibody was conjugated with DTPA and radiolabeled with 111 In for imaging, and commercialized under the name ProstaScint® (Cytogen Corporation). 121 A huJ591 mAb was radiolabeled with 177Lu and evaluated in clinical trials in subjects with mCRCP. 122 Recently, PSMA-617, a ligand with optimal pharmacokinetics, was conjugated with a DOTA chelator for PSMA-targeted radioligand therapy. 123
Nevertheless, some patients fail to respond to 177Lu-labeled PSMA ligands, and despite the good tolerability, the long path length of β− particles continues to be a risk factor. 124
This opens an opportunity for α radiation therapy labeled to PSMA ligands. Kratochwil et al. took this opportunity to evaluate PSMA-targeted α radiation therapy with 225Ac-PSMA-617 in a first-in-human trial, in one case of contraindication for treatment with β− emitters and another where the patient failed to response to 177Lu-PSMA-617 therapy. These patients experienced a remarkable benefit from TAT. 104
Conclusions
TAT allows selective delivery of highly toxic radiation to target cells with reduced harm to surrounding cells. Because of their high LET and short path length, α-emitting radionuclides offer a number of competitive advantages over other therapeutic radionuclides, including higher RBE; reduced dependence of radioresistance mechanisms due to increased direct DNA damage; and reduced toxicity due to their limited range. However, the widespread acceptance of α-emitting radionuclides for clinical applications has been obstructed by low availability of radionuclides and concerns about potential toxicity due to free daughter radionuclides.
Although nonuniform dose distribution among target cells is still a significant limiting factor, there is now evidence that TAT can promote tumor regression and stimulate the immune response. Building on this concept, the complexity of therapeutic radiopharmaceuticals is changing from a simple radioactive element such as 223Ra to a conjugated carrier molecule (i.e., 225Ac-PSMA-617). This opens an opportunity to extend the application of TAT to other malignant diseases. Obviously, there are many challenges still ahead at this stage.
Current research into α-emitting radiopharmaceuticals is mainly focused on the development of more stable and selective carriers. The urgent need for more specific binding was highlighted by some challenges with the use of 223RaCl2, whose nonspecific attraction to actively growing bone leads to uptake on healthy bone tissue.
Consequently, TAT-specific radiobiology also needs to be studied. It is essential to clarify the contribution of indirect effects to the lethality of α-emitting radiopharmaceuticals, taking into account their unique microdosimetry. It is starting to be widely accepted that biological effects of TAT extend beyond the range of α particles, due the bystander effects, stimulation of immune system and TAVAT, which could fundamentally change how TAT is planned.
Also, there is a need to conduct further controlled randomized trials with sufficient patient numbers to be able to properly compare and evaluate different radionuclides and conjugating agents. It is clear that TAT is far from its full potential and using body weight to calculate the administered activity may not be the optimal parameter.
A greater understanding of the radiobiology of high LET α particles can lead to the design of safer and more efficient approaches for the delivery of α particle radiation. Advances in TAT imaging and dosimetry will provide the fundamental knowledge for optimization and treatment planning. With regard to TAT, labeled 227Th and 225Ac seem to be promising and could represent the next approaches to extend survival in the future for several clinical settings, alone or in combination with other treatments. However, the current supply of 227Th and 225Ac is certainly insufficient for routine applications in hospitals worldwide and developing new supply chains will be critical.
Footnotes
Authors' Contributions
F.D.C.G.L.: Investigation, writing—original draft preparation, and writing—review and editing; S.J.M.: Writing—review and editing, and conceptualization; J.M.O: Writing—review and editing; K.M.P.: Writing—review and editing, supervision, and funding acquisition.
Disclaimer
All authors of this article have reviewed and approved it before submission.
Disclosure Statement
F.D.C.G.L., and S.J.M. declare no conflict of interests regarding the publication of this article. J.M.O. has received honoraria for Speakers bureau and Advisory Board from Bayer. J.M.O. has received institutional research funding from Bayer. K.M.P. has received speaker honoraria from Bayer.
Funding Information
The authors gratefully acknowledge the support of Fundação para a Ciência e Tecnologia (FCT-MCTES), Radiation Biology and Biophysics Doctoral Training Programme (RaBBiT, PD/00193/2012); Applied Molecular Bioscience Unit (UCIBIO) (UIDB/04378/2020) and Centre of Physics and Technological Research (CEFITEC) Unit (UIDB/00068/2020); and scholarship grant number SFRH/BD/114448/2016 to (F.D.C.G.L.). The work was funded by the Movember Prostate Cancer United Kingdom Centre of Excellence (CEO13_2-004) and the Research and Development Division of the Public Health Agency of Northern Ireland (COM/4965/14).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.