
Abstract
Prostate cancer is the most common cancer to affect men in the United States and the second most common cancer in men worldwide. Prostate-specific membrane antigen (PSMA)-based positron emission tomography (PET) imaging has become increasingly popular as a novel molecular imaging technique capable of improving the clinical management of patients with prostate cancer. To date, several 68Ga and 18F-labeled PSMA-targeted molecules have shown promising results in imaging patients with recurrent prostate cancer using PET/computed tomography (PET/CT). Studies of involving PSMA-targeted radiopharmaceuticals also suggest a higher sensitivity and specificity, along with an improved detection rate over conventional imaging (CT scan and methylene diphosphonate bone scintigraphy) and 11C/18F-choline PET/CT. In addition, PSMA-617 and PSMA I&T ligands can be labeled with α- and β-emitters (e.g., 225Ac, 90Y, and 177Lu) and serve as a theranostic tool for patients with metastatic prostate cancer. While the clinical impact of such concept remains to be verified, the preliminary results of PSMA molecular radiotherapy are very encouraging. Herein, we highlighted the current status of development and future perspectives of PSMA-targeted radiopharmaceuticals and their clinical applications.
Introduction
Prostate cancer is the most common cancer to affect men in the United States 1 and the second most common cancer in men worldwide. 2 Diagnosis and treatment of primary prostate cancer have vastly improved with advances in surgical resection, radiotherapy, hormonal therapy, and chemotherapy. Initial diagnosis typically involves cancer detection through digital rectal examination and an elevation of serum prostate-specific antigen (PSA) levels, followed by ultrasound-guided biopsy of the prostate gland. 3 Once the diagnosis of prostate cancer is established, treatment is selected according to the anatomic extent of disease (tumor, node, metastasis stage), Gleason score/tumor grade of biopsy samples, number and extent of tumor involvement in the biopsy specimens, pretreatment serum PSA levels, and more recently genomic profiling. Imaging studies can also be used in selective patients, according to the initial risk stratification. Even though various therapeutic strategies are currently available, optimized treatment can only be achieved through the precise identification of patients with truly localized versus metastatic disease. However, conventional imaging techniques such as computed tomography (CT) and bone scintigraphy used for initial staging and restaging after cancer recurrence have limited sensitivity. More recently, several new radiotracers for positron emission tomography (PET)/CT imaging of prostate cancer have become clinically available in the United States. 4 Nevertheless, identifying sites of active disease remains challenging when the tumor burden is low; therefore, new and more sensitive imaging techniques are warranted.
Prostate-specific membrane antigen (PSMA)-based PET imaging is emerging as an important tool for the management of prostate cancer patients. Moreover, PSMA radioligands can be modified to deliver systemic targeted radiotherapy to tumor cells and serve as a theranostic tool in the evaluation of post-therapy response. The aim of this article is to provide an overview of the recent advances in PSMA-targeted radiopharmaceuticals and their application in initial staging and restaging of prostate cancer. In addition, as some of these radiopharmaceuticals can also be used to predict response of PSMA-targeted therapy through real-time in vivo assessment of PSMA expression, the use of PSMA constructs as theranostic agents will also be discussed.
Antibody-Based Radiopharmaceuticals in PSMA-Targeted Imaging and Therapy
PSMA is a type II transmembrane glycoprotein that comprises (1) a 19-amino-acid internal portion, (2) a 24-amino-acid transmembrane portion, and (3) a 707-amino-acid external portion. 5,6 It was originally characterized by the murine monoclonal antibody (mAb) 7E11-C5.3. 7 Although lower level of PSMA expression can be observed in normal tissues other than prostate such as kidney, liver, small intestine, and brain, 8,9 PSMA is highly expressed in prostate cancer cells and in nonprostatic solid tumor neovasculature. 10 In fact, PSMA is reported to be overexpressed 100–1000 fold in 95% of prostate cancer cells. 11
As 7E11 is the first anti-PSMA antibody that recognizes and binds a PSMA intracellular or cytoplasmic epitope, 111 In-labeled 7E11 (ProstaScint™) has been investigated clinically for imaging recurrent and metastatic prostate cancer after approval by the U.S. Food and Drug Administration in 1996 as a single photon emission computed tomography (SPECT) radiopharmaceutical. 12,13 The overall sensitivity and specific activity of ProstaScint is 60% and 70%, respectively. 14 –16 However, since PSMA intracellular epitopes are only accessible during the membrane disruption process in dead, dying, or apoptotic cells, the use of ProstaScint for clinical diagnosis is limited. 17 Consequently, a humanized mAb huJ591 (J591) that targets to the extracellular domain of PSMA was developed for imaging and therapy of prostate cancer.
J591 has been studied extensively in preclinical models and demonstrated excellent tumor binding specificity in the prostate cancer xenografts. 18 According to the recent phase I/II study of 89Zr (T 1/2: 78.4 h; β+: 23%; Eβ + max: 0.90 MeV) labeled J591 as a PET imaging agent for metastatic prostate cancer, the overall accuracy of 89Zr-J591 was 95.2% for osseous lesions and 60% for soft-tissue lesions. 19 While 89Zr-J591 PET is comparable to CT and 18F-FDG PET in monitoring soft-tissue lesions, 89Zr-J591 imaging demonstrates superior targeting of bone lesions.
J591 is also currently the only humanized mAb used to targeted radiation therapy directly to the tumor cells. 20 Although it was initially labeled with 213Bi and evaluated as a promising targeted α-particle therapeutic agent, 21 the limited availability of the 225Ac/213Bi generator system hampers the further application of 213Bi-J591 in clinical use. As a result, J591 was labeled with 90Y or 177Lu for clinical evaluations. Based on the clinical trials performed in Cornell University, the maximum tolerated dose (MTD) of 177Lu-J591 in a single dose is significantly higher compared with 90Y-J591 (2590 vs. 647.5 MBq/m2). 22 The MTD of 177Lu-J591 can further be increased to 3330 MBq/m2 when applying dose fractionation (two doses apart of 1665 MBq/m2, 2 weeks apart). 22 Although myelosuppression is the dose limiting toxicity, it can be overcome through dose fractionation or in combination with other chemotherapeutic agents to stabilize the progression of prostate cancer. 23 However, because small molecules are advantageous in faster blood clearance and better tumor permeability over much larger constructs such as monoclonal antibodies, the focus of current investigative efforts is therefore switched to develop PSMA inhibitor-based radiopharmaceuticals, as they are typically small molecular agents.
Advancement of PSMA Inhibitor-Based Radiopharmaceuticals in Imaging Prostate Cancer
The enzymatic activity of PSMA is considered part of the catalytic zinc metallopeptidase family M28. Although PSMA is encoded by folate hydrolase 1 gene (FOLH1) that reflects its major role in folate uptake, PSMA usually acts as a glutamate carboxypeptidase II in different tissues and hydrolyzes N-acetylaspartylglutamate (NAAG). 24,25 Therefore, derivatives based on NAAG were developed as PSMA inhibitors and evaluated for their targeting capabilities.
Phosphorous-based compounds such as 2-phosphonomethyl pentanedioic acid and 4,4′-phosphinicobis(butane-1,3-dicarboxylic acid) were the first designed ligands with high affinity toward PSMA <1 nM. 26,27 However, due to the high polarity and relatively poor pharmacokinetic profiles of these compounds, their clinical applications are limited. 28
Several variants of the thiol-, hydroxamate-, and sulfonamide-containing compounds were subsequently evaluated as an alternative to phosphorus-containing molecules as they demonstrated enhanced membrane permeability and oral bioavailability. 29 –32 However, their clinical applications are hampered by their relatively low PSMA selectivity and metabolic stability. 28 To overcome the limitations that have occurred in the aforementioned compounds, extensive structure-activity studies were performed and yielded to a series of urea-based ligands. Many of these urea-based ligands are found to be highly specific toward PSMA, thus have been further applied for diagnostic and therapeutic purposes.
11C-MCG/11C-DCMC
11C-MeCys-C(O)-Glu (11C-MCG/11C-DCMC) (Fig. 1) is the first urea-based radiotracer that targets PSMA. Although this compound was initially reported in 2002, 33 its preclinical evaluation for imaging prostate cancer was not revealed until 2005. 34 The biodistribution of 11C-MCG/11C-DCMC was conducted in the mice bearing with MCF-7 (breast, PSMA-negative), PC-3 (prostate, PSMA-negative), and LNCaP (prostate, PSMA-positive) xenografts. The tracer showed high tumor-to-muscle (T/M) ratio in LNCap-derived tumors (10.78) and significantly low T/M ratios in MCF-7- and PC3 counterparts (1.28 and 2.10, respectively). The biodistribution aligned with the small animal PET imaging results and these encouraging data were further initiated an extensive research on urea-based radiopharmaceuticals in PSMA-targeted imaging and therapy.

Chemical structures of PSMA inhibitor-based radiopharmaceuticals. PSMA, prostate-specific membrane antigen.
123I-MIP-1072 and 123I-MIP-1095
Sodium [123I]iodide is a radiotracer used for SPECT or SPECT/CT scans with a half-life of 13.2 h. Molecular Insight Pharmaceuticals, Inc. (acquired by Progenics Pharmaceuticals, Inc. in 2013) initiated a program in 2006 to develop a series of radio-conjugates for prostate cancer imaging and therapy. 35 123I-MIP-1072 and 123I-MIP-1095 (Fig. 1) are the two lead compounds that showed high PSMA binding affinity. The pilot clinical studies of 123I-MIP-1072 and 123I-MIP-1095 demonstrated these two radiotracers as useful PSMA-targeting SPECT agents. 36 Derived from this promising result, the phase I clinical study of 131I-MIP-1095 has recently been reported regarding its therapeutic efficacy in men with metastatic castration-resistant prostate cancer, in which a PSA decline of ≥50% was achieved in 70.6% of the patients (total patient number: 36) from the initial treatment. However, after the initial treatment, the group observed that 131I-MIP-1095 became less effective and resulted in more frequent and intense side-effects to the patients, especially hematologic toxicities and xerostomia. 37
99mTc-MIP-1404 and 99mTc-MIP-1405
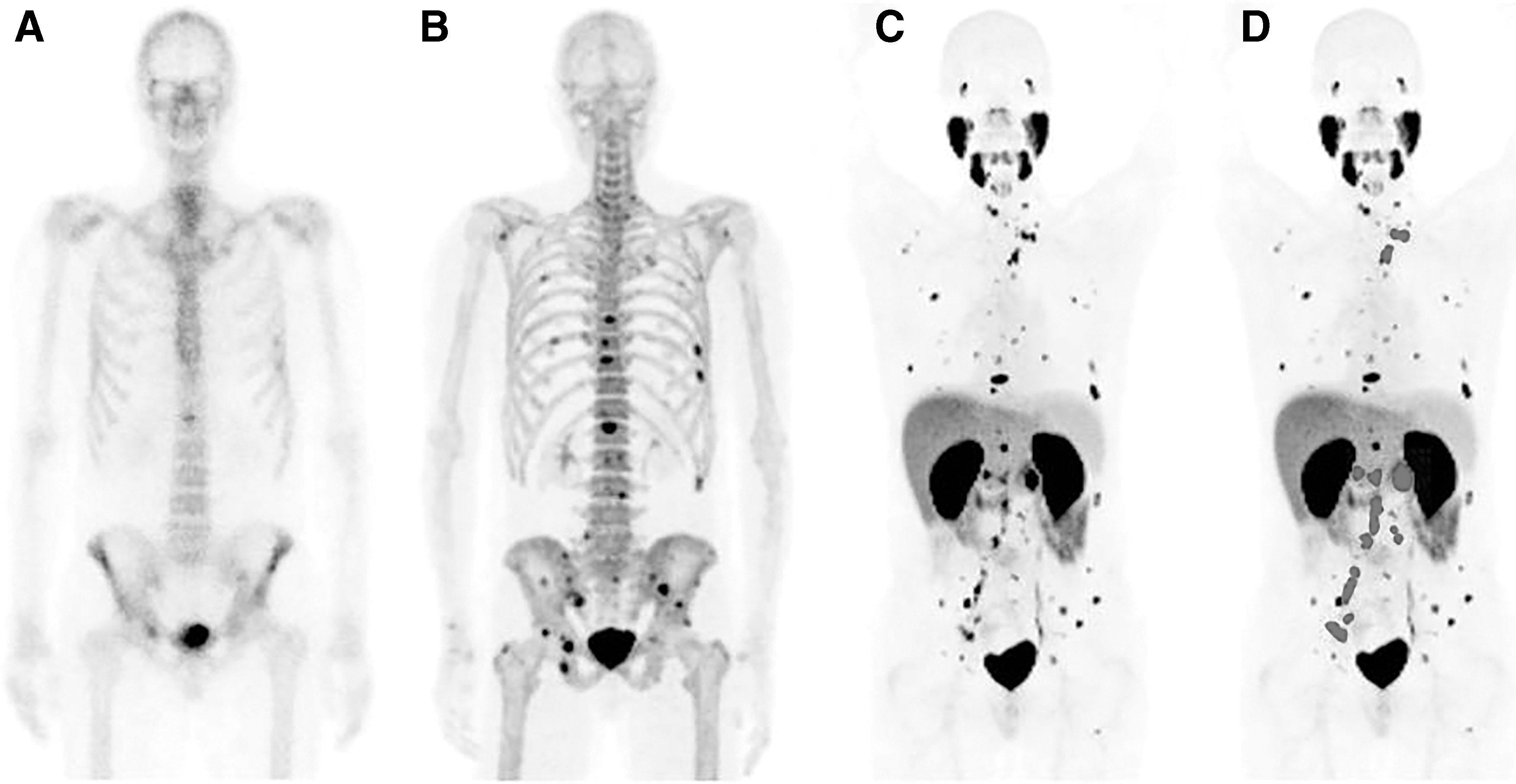
Technetium-99m (99mTc) is the most commonly used SPECT isotope with a half-life of 6 h. Although multiple urea-based PSMA ligands labeled with 99mTc have been developed in the past decade, 38 –41 only 99mTc-MIP-1404 and 99mTc-MIP-1405 (Fig. 1) were clinically evaluated. 42,43 According to the pilot SPECT scans of 99mTc-MIP-1404 and 99mTc-MIP-1405 in patients with metastatic prostate cancer, Vallabhajosula et al. observed that both tracers are able to identify most metastatic lesions in bone and soft tissues (Fig. 2). 43 However, because 99mTc-MIP-1404 has relatively low accumulation in the bladder, its potential for staging and monitoring treatment response in patients with prostate cancer was further evaluated.
Whole-body planar images of 99mTc-MIP-1404
A recent clinical study conducted by Schmidkonz et al., which involved 93 patients with histologically confirmed prostate cancer, showed an overall positive 99mTc-MIP-1404 SPECT/CT detection rate of 97%. In addition, interobserver agreement was high for the overall scan result (97%), as well as for the detection of the primary tumor (97%), of lymph node metastases (97%), and of bone metastases (99%). 42 When applying 99mTc-MIP-1404 SPECT/CT to assess the treatment response in patients with metastatic prostate cancer through the same research group, its concordance rate to biochemical response based on the change of PSA levels was found to be 75%. 44 These encouraging results suggest a possible role of 99mTc-MIP-1404 SPECT/CT for staging and monitoring treatment in patients with prostate cancer.
68Ga-PSMA-11
Gallium-68 (68Ga) is a short-lived radionuclide (T 1/2: 68 min) and primarily decays through positron emission (87.94%) with a maximum energy of 1.9 MeV (mean energy: 0.89 MeV). When Obninsk first made a commercially available 68Ge/68Ga generator that could be eluted by dilute hydrochloric acid to provide cationic 68Ga in 1996, 45 it opened new possibilities of 68Ga in preclinical and clinical applications. As cationic 68Ga is able to form stable complexes with chelators (e.g., diethylenetriaminepentaacetic acid, 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid [DOTA], etc.) that have already been widely used to the agents for magnetic resonance imaging (MRI) and SPECT, the amount of 68Ga related research has greatly increased since 2000, especially after the first successful clinical trial of 68Ga-DOTATOC in 2001. 46
Among all PSMA inhibitor-based radiopharmaceuticals, 68Ga-PSMA-11 (Fig. 1) is perhaps the most investigated PET agent for imaging prostate cancer. Afshar-Oromieh et al. initiated the evaluation of 68Ga-PSMA-11as a novel PET agent for prostate cancer. 47 The researchers found that 68Ga-PSMA-11 detects recurrent and metastatic prostate cancer through binding to the extracellular domain of PSMA and through internalization of the compound/agent into the cell. Since then, multiple studies have been conducted to compare 68Ga-PSMA-11 with other conventional PET tracers for imaging patients with prostate cancer.
A recent prospective study conducted by Caroli et al., 48 which involved 314 patients with recurrent prostate cancer, showed an overall positive 68Ga-PSMA-11 PET/CT detection rate of 62.7%. The accuracy was increased with increasing PSA values: 42% for 0–0.2 ng/mL, 58% for 0.2–1.0 ng/mL, 75% for 1.0–2.0 ng/mL, and 94.8% for >2.0 ng/mL. In addition, the researchers found that of the 88 patients with negative fluorine 18F-choline PET/CT scans, 59 (67%) were positive on 68Ga-PSMA-11 PET/CT. Of these positive scans, 57% had a PSA value <2.0 ng/mL and 81% had a Gleason score of ≥7. In agreement to these findings, a positive 11C-choline PET/CT detection rate was also reported with poor correlation when patients had a PSA value <2.0 ng/mL. 49
With encouraging results continuously shown in prospective studies and meta-analysis, 2,48 –54 several analogs based on PSMA-11 have also been evaluated as potential imaging agents for prostate cancer. 55 –59 Among these molecules, PSMA-617 (Fig. 1) has received the most clinical attention. Through preclinical studies, Benesova et al. observed that the binding affinity of PSMA-617 is significantly improved toward PSMA (Ki for PSMA-11: 12 ± 2.8 nM, PSMA-617: 2.3 ± 2.9 nM), as well as its internalized ratio into prostate cancer cells (internalized ratio for PSMA-11: 9.47% ± 2.56% injected activity/106 LNCaP cells, PSMA-617: 17.67% ± 4.34% injected activity/106 LNCaP cells). 60 As DOTA is used as a chelator for PSMA-617, this tracer can be labeled with 68Ga, 111 In, 177Lu, 90Y thus served as a theranostic agent; however, when comparing with 68Ga-PSMA-11, although both tracers showed the highest uptake in the kidneys and salivary glands, 57,61 the pharmacokinetics of 68Ga-PSMA-617 is slower in patients. Because most nuclear medicine clinical workflow is designed to conduct PET/CT scans within 1 h after injection of 68Ga-labeled tracers, it remains unclear if 68Ga-PSMA-617, even with its higher PSMA binding affinity and internalization nature, could detect more prostate cancer lesions than 68Ga-PSMA-11. Therefore, 68Ga-PSMA-11 with its putatively faster clearance provides a clear advantage over 68Ga-PSMA-617.
In addition to imaging patients with recurrent prostate cancer, 68Ga-PSMA-11 PET/CT can also be a valuable tool for evaluating primary prostate cancer and detecting lymph node and bone metastases. Kabasakal et al. imaged 28 prostate cancer patients with 68Ga-PSMA-11 PET/CT either 5 or 60 min postinjection (p.i.) and found that images acquired on the early time point (5 min p.i.) can help better distinguish between urinary bladder and tumor lesions. 62 Perveen et al. further confirmed this observation by performing dynamic 68Ga-PSMA-11 PET/CT in 15 prostate cancer patients within a time frame of 1–10 min and then compared these images to the static ones that were acquired between 45 and 60 min after injection from the same patients. 63 In their own experience, the authors observed that 68Ga-PSMA-11 PET/CT occasionally reveals metastases from prostate cancer to unusual locations, such as the skin (Fig. 3). In this case, their images demonstrate a tiny 3 mm skin lesion in the perineum, which explained the patient's rising PSA levels. After the metastasis was removed, the PSA normalized. This tiny metastatic lesion was confirmed by histology and PSA-immunohistochemical staining.
68Ga-PSMA-11 imaging at 15 min (upper row) and at 60 min (lower row). This 67-year-old man had originally Gleason Score 8 prostate cancer, treated initially with transurethral resection of the prostate, androgen deprivation, and external beam radiation therapy, but developed 4 years later mediastinal lymph node metastases. The patient was then treated with docetaxel and abiraterone, but 2 years later rising PSA was detected (PSA was 3.1 ng/dL). A 3 mm perineum metastasis was identified, better depicted in the lower row images (arrows). It was surgically removed; histology (upper right image) revealed malignant histology and immunohistochemistry (lower right image) with PSA staining proved. PSA, prostate-specific antigen.
18F-DCFBC, 18F-DCFPyL, and 18F-PSMA-1007
Fluorine-18 (18F) is the most widely used PET isotope. This is because 18F can be produced through a medical cyclotron (9–20 MeV) with a great amount of activity and its excellent decay characteristics (T 1/2: 109.7 min, β+ branch ratio: 97%, E max: 0.633 MeV). The 18F-labeled PSMA inhibitors start to receive great attention after 18F-DCFBC and 18F-DCFPyL (Fig. 1) were reported.
18F-DCFBC is structurally similar to 11C-DCMC and was initially reported in 2008. 64 In 2012, its pilot clinical PET/CT studies were performed in 5 patients with radiological evidence of prostate cancer. 65 According to the report, 32 PET-positive suspected metastatic sites were identified, with 21 concordant on both PET and conventional imaging for abnormal findings compatible with metastatic disease. Of the 11 PET-positive sites not identified on conventional imaging, most were within the bone and could be considered suggestive for the detection of early bone metastases. In a comparison of 18F-DCFBC with 99mTc-methylene diphosphonate (MDP) bone scan and contrast-enhanced CT of the chest, abdomen, and pelvis in 17 patients, 18F-DCFBC PET/CT has better sensitivity and was superior to detect lymph nodes, bone lesions, and visceral lesions than these imaging modalities. 66 With these encouraging clinical findings, the correlation of 18F-DCFBC PET/CT with multiparametric MRI (mpMRI) and histopathology has recently been investigated. 67 However, although 18F-DCFBC PET/CT has a higher positive predictive value suggesting a potential role in patients at high risk for biopsy, relatively low sensitivity (36%) of 18F-DCFBC PET/CT was observed compared with mpMRI (96%) in detecting all tumor lesions at 13 patients.
Following the clinical success of 18F-DCFBC, 18F-DCFPyL is the next generation of 18F labeled PSMA inhibitor-based radiopharmaceutical. Although the design of both tracers is based on Lys-urea-Glu pharmacophore, the 18F is introduced through a fluorinated pyridine derivative that is connected to Lys to yield a more stable amide bond with the pharmacophore. Compared to 18F-DCFBC, 18F-DCFPyL is reported to have superior binding affinity toward PSMA (Ki for 18F-DCFBC: 13.5 nM; Ki for 18F-DCFPyL: 1.1 nM). 64,68 Based on the initial evaluation of 18F-DCFPyL in 9 patients with prostate cancer, its biodistribution profile is superior to that observed with 18F-DCFBC, exhibiting higher tumor-to-blood and tumor-to-muscle contrasts. 69 Furthermore, a secondary analysis of 8 patients in this initial clinical trial showed that a markedly increased number of lesions were visible on 18F-DCFPyL PET/CT in comparison to conventional imaging with CT and MDP bone scan. When taking into account intra-patient clustering effects through use of a general estimating equation regression model, it was estimated that a proportion of 0.72 of the lesions that would be negative or equivocal with CT and MDP bone scan would be definitively positive with 18F-DCFPyL PET/CT, whereas only an estimated proportion of 0.03 of the lesions that would be negative or equivocal with 18F-DCFPyL PET/CT would be definitively positive on conventional imaging. These findings were in agreement with a case report comparing 18F-DCFPyL PET/CT with 18F-NaF PET/CT and MDP bone scans in a patient with extensive bone metastatic prostate cancer, in which several putative sites of disease were only revealed by 18F-DCFPyL PET/CT (Fig. 4). 70 In recently reported clinical trial, a total of 248 consecutive patients with biochemically recurrent prostate cancer were evaluated and underwent scanning with 18F-DCFPyL PET/CT. Scan positivity is correlated with the PSA values: 17/29 scans (59%) with PSA values <0.5 ng/mL; 20/29 (69%) with PSA 0.5 to <1.0 ng/mL; 35/41 (85%) with PSA 1.0 to <2.0 ng/mL; 69/73 (95%) with PSA 2.0 to <5.0 ng/mL; and 73/76 (96%) with PSA ≥5.0 ng/mL. These results appear to be comparable to those reported for 68Ga-PSMA-11, with potentially increased detection efficacy compared to 68Ga-PSMA-11 for patients with PSA <2.0 ng/mL. 71
In addition to 18F-DCFBC and 18F-DCFPyL, 18F-PSMA-1007 (Fig. 1) is another promising tracer that has recently been introduced clinically. 72 According to the meta-analysis by Treglia et al., 73 18F-PSMA-1007 and 18F-DCFPyL have similar detection rate in patients with biochemical recurrent prostate cancer. However, nonurinary excretion of 18F-PSMA-1007 might be advantageous over 68Ga-PSMA-11 and 18F-DCFPyL in cases of local recurrence and unclear lesions in proximity to the ureter or urinary bladder (Fig. 5). 74,75
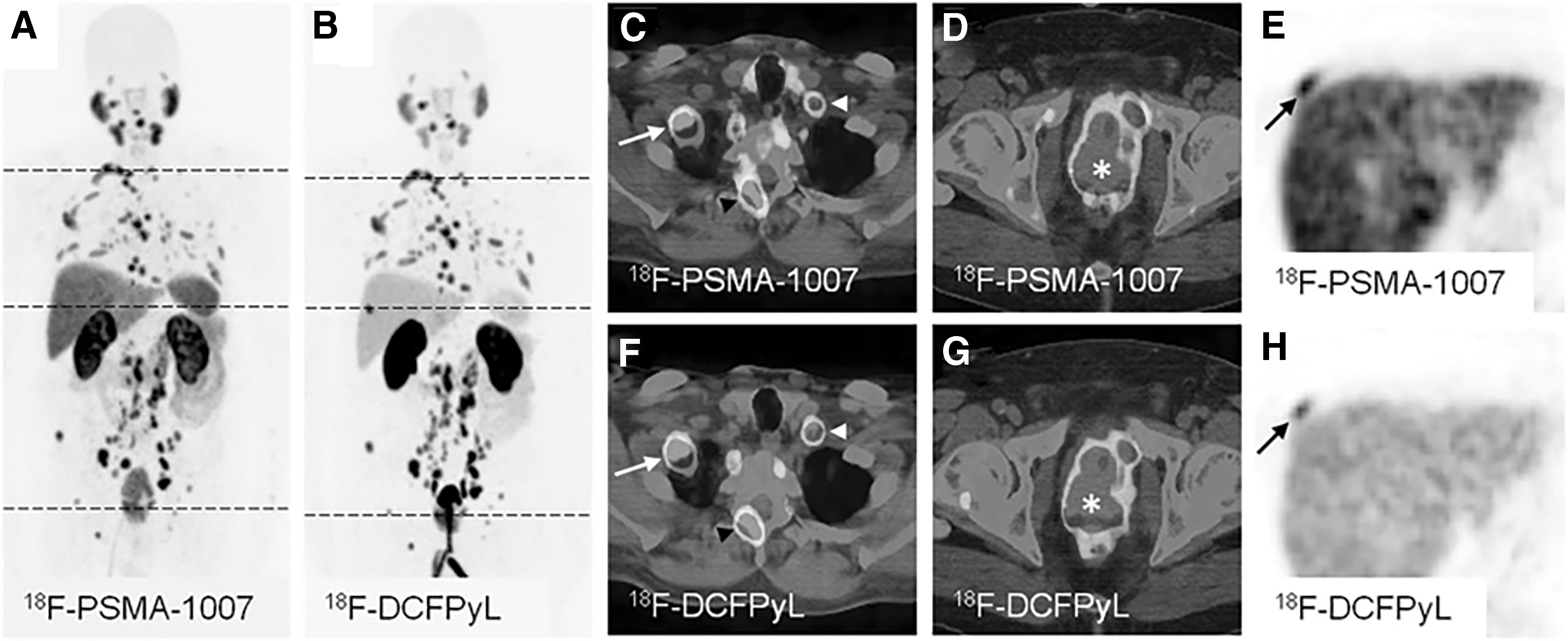
Maximum-intensity projections of PET examinations using 18F-PSMA-1007
PSMA Molecular Radiotherapy
Interest in PSMA molecular radiotherapy (MRT) has been greatly increased with the recent success of the 177Lu-DOTATATE (LUTATHERA®) trial showing markedly longer progression-free survival and a significantly higher response rate than high-dose octreotide of long-acting release among patients with advanced midgut neuroendocrine tumors. 76 The goal of MRT is to precisely deliver as much radiation as possible to the cancer cells but with minimal effect in the normal organs/tissues. With continuous advancement of PSMA-targeted radiotracers, theranostic agents which share the same constructs are now under investigation for the treatment of metastatic castration resistant prostate cancer.
177Lu-PSMA-617 is currently most studied PSMA MRT agent, and its first retrospective multicenter cohort study comprising 145 patients from 12 centers in Germany has been published in 2017. 77 These patients received up to four MRT cycles of 177Lu-PSMA-617 (range of administered activity 2–8 GBq; 5.9 GBq in average). After a single treatment, 66% of the patients experience the decrease of PSA, and a PSA decrease of more than 50% was observed in 40% of the patients. Side-effects, including mild hematotoxicity, as well as mild but transient xerostomia, were found in 10% and 8% of the patients, respectively. The promising response rates and a low toxicity nature of 177Lu-PSMA-617 have also been confirmed by other recent clinical evaluations. 78 –83
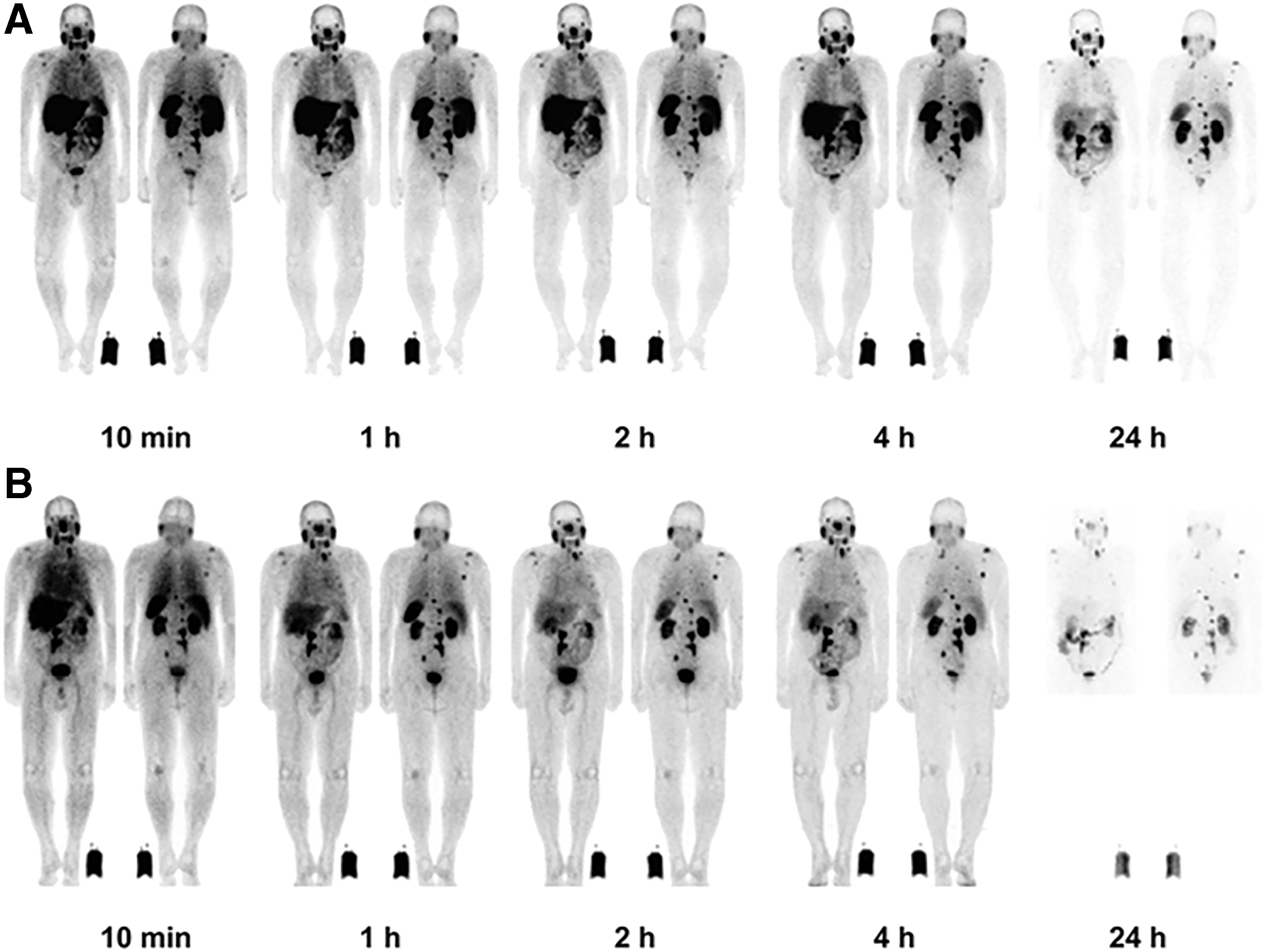
In addition to 177Lu-PSMA-617, the therapeutic efficacy of 177Lu-PSMA I&T (Fig. 6) has received great attention in recent years as well. According to the clinical report from Kulkarni et al., although both radiopharmaceuticals share similar biodistribution profiles, the whole-body dose was moderately higher for 177Lu-PSMA-617. 84 Like 177Lu-PSMA-617, very little toxicity has been observed in clinical routine practice in European centers. 85 Figure 7 demonstrates the time-activity behavior using a therapeutic dose of 177Lu-PSMA I&T and whole-body imaging at 0.5, 4, 24, and 168 h. The targeting signal increases throughout the study.
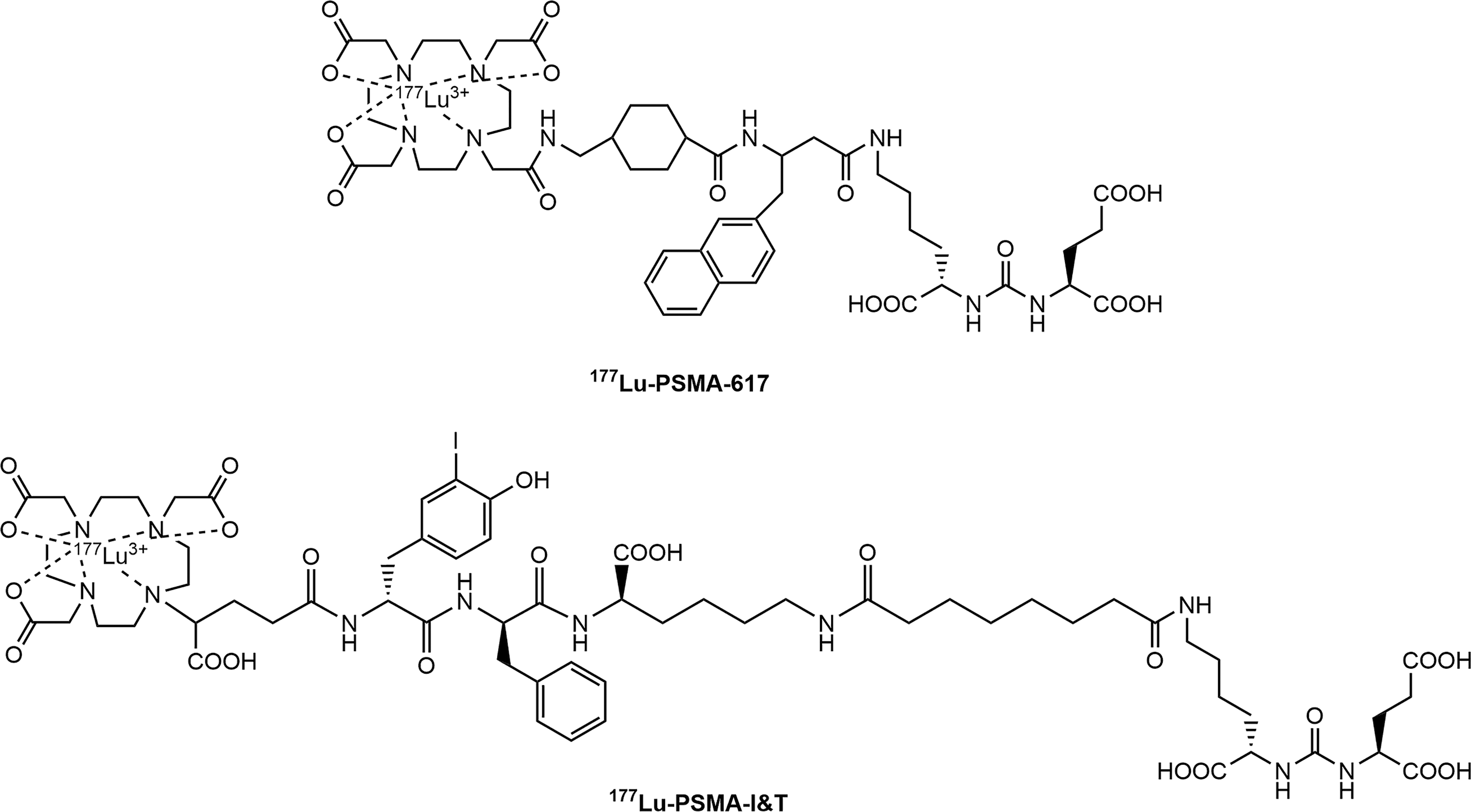
Chemical structures of 177Lu-PSMA-617 and 177Lu-PSMA I&T.
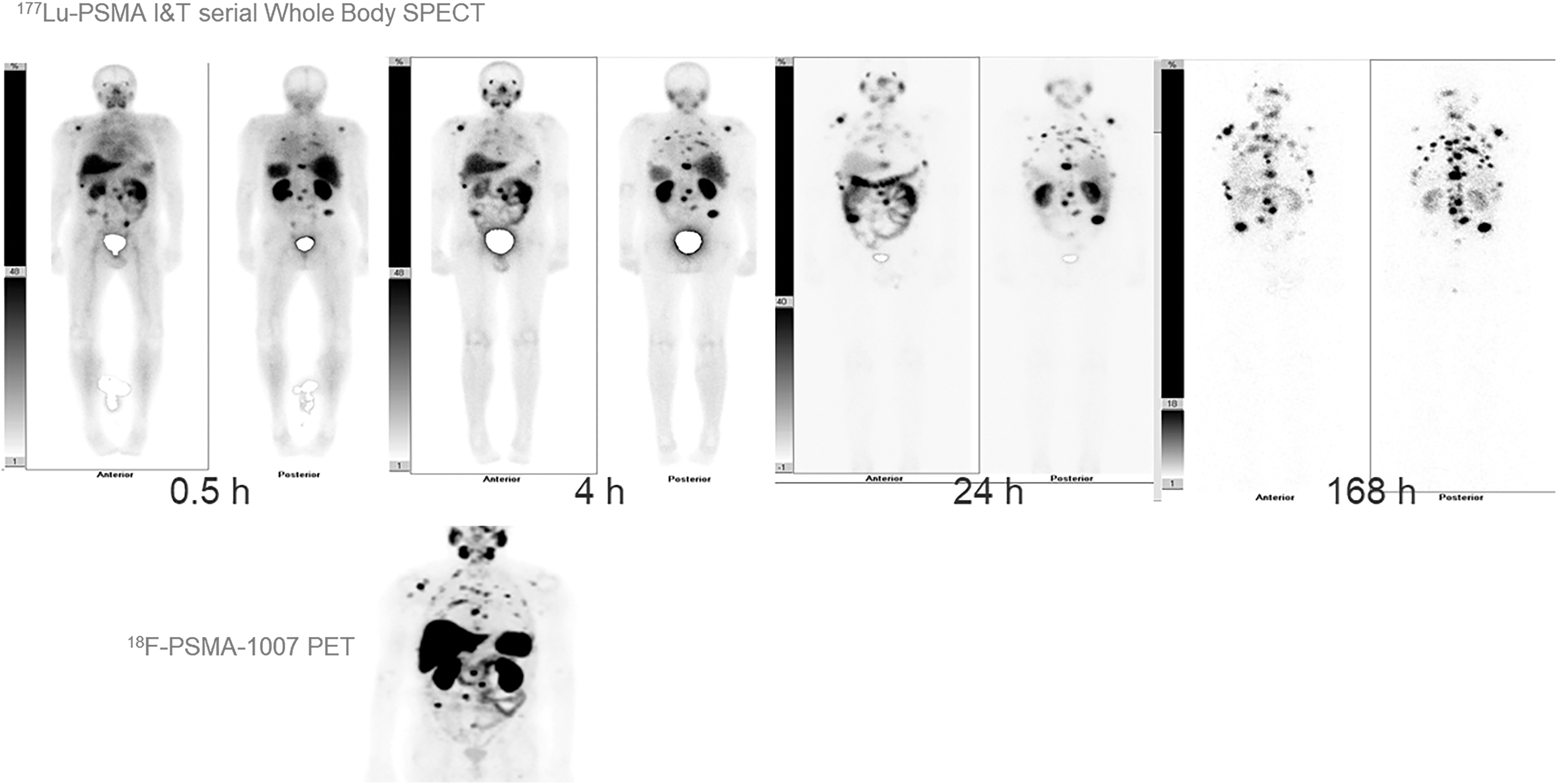
177Lu-PSMA-I &T serial whole body at 0.5, 4, 24, and 168 h in a widespread advanced prostate cancer (bone and lymph node disease), AP/PA views. The diagnostic pretherapeutic 18F-PSMA-1007 PET image is shown for comparison.
Although 177Lu-PSMA-617 and 177Lu-PSMA I&T studies have shown convincing therapeutic response in terms of both serum PSA level and radiologic findings, about 30% of patients were not responsive and dose escalation was severely limited by chronic hematological toxicity. 77,86 As a result, targeted α-therapy (TAT) that could potentially prevent radioresistance to β-emitters such as 177Lu and reduce hematological toxicity has recently received great attention. The PSMA TAT was initiated by Kratochwil et al. with the administration of 225Ac-PSMA-617 in 2 patients. 87 The PSA of both patients declined to undetectable levels (<0.1 ng/mL), and lesions showed a complete response on imaging. With this encouraging result, 225Ac-PSMA-617 has further been investigated in 17 chemotherapy-naive patients leading to a ≥90% decline in serum PSA in 82% of patients. Importantly, 41% of these patients remained in remission 12 months after therapy. 88 While xerostomia was the primary side-effect, no severe hematologic toxicity was observed. Therefore, these preliminary results warrant further clinical evaluations.
Perspectives of Research and Development
Use cyclotron-produced 68Ga for 68Ga radiopharmaceuticals
The dramatic rise in PET imaging with 68Ga-labeled tracers during the last decade has been a result of increased interest in applying 68Ga radiopharmaceuticals for theranostic purposes. The theranostic concept is when a diagnostic scan is first performed using a 68Ga-labeled ligand then the same ligand is labeled with a therapeutic radionuclide (e.g., 90Y, 177Lu, or 188Re) for treatment. 89 –91 Currently, 68Ga is commonly provided through 68Ge/68Ga generators. Although high production volumes of the 68Ge/68Ga generators have recently been made to meet existing needs, there is a strong commercial appeal for cyclotron-produced 68Ga to overcome limited availability of 68Ga radiopharmaceuticals, particularly in densely populated regions. Consequently, the development of an alternative way to meet the increasing demand for 68Ga is warranted.
Several attempts have been made to produce 68Ga via 68Zn(p,n)68Ga reaction using a cyclotron. Jensen and Clark reported the first attempt to produce 68Ga using a cyclotron with a liquid target filled with a 68ZnCl2 solution. 92 Since then, other groups have tried to optimize 68Ga production through liquid targetry. 93,94 Based on recent study performed by Alves et al., a liquid targetry containing 200 mg of 68Zn resulted in 0.3 GBq/μA · h at the end of bombardment when the target was irradiated with 14.2 MeV protons. 93 Overall, producing 68Ga using a liquid target is advantageous in minimizing radiation exposure during the target handling and processing. However, the available activity of 68Ga from this technique is not significantly higher than activity obtained through a generator. In addition, the liquid target is with risk of excessive pressure during irradiation and requires more maintenances in the target and transfer lines that may limit its use for routine clinical productions. 94,95
The possibility of producing 68Ga with a solid target was first explored by Engle et al. using natZn as the target material. 96 Derived from cross-section measurements, the theoretical production yield can be up to 5.81 GBq/μA · h when an enriched 68Zn solid target is irradiated by 15 MeV proton 97 ; however, high zinc and HCl acid contents in the final 68Ga solution are two potential challenges for cyclotron-produced 68Ga. Although most 68Zn can be removed using the AG50W cation resin alone, 93,94,96 additional processing is usually required to reconstitute 68Ga solution with a low concentration of HCl for radiolabeling use. Considering the relatively short half-life of 68Ga, a simple, fast, and cost-efficient processing procedure of 68Ga post target irradiation is required.
In the Cyclotron Radiochemistry Facility at MD Anderson Cancer Center, the authors have developed a reliable and cost-efficient procedure for cyclotron produced 68Ga, using solid target and chemical separation that are high in quality and yield. 98 In response to the need for a more efficient and economically viable alternative and to explore the potential applications of cyclotron-produced 68Ga in routine radiopharmaceutical preparation, the authors have developed a simple and fully automated method of producing cGMP 68Ga-PSMA-11 using cyclotron-produced 68Ga and verified its biological equivalence. Preliminary data indicate the potential for large scale production of 68Ga radiopharmaceuticals that can meet the increasing demand and facilitate regional distribution of these imaging agents. 99 Current development of 68Zn pressed target is underway to reduce time for target preparation and further establish a more efficient way to produce 68Ga on a routine daily basis.
Apply Al18F as an alternative 18F-labeling technique for PSMA-targeted molecules
Due to longer half-life of 18F (t 1/2 = 109.8 min) allowing both imaging studies over a few hours and easier local distribution, radiolabeling PSMA molecules with 18F represents an attractive alternative to its 68Ga-based counterpart. In fact, recent clinical trials of both 18F-DCFPyL and 18F-PSMA-1007 have exhibited excellent sensitivity for the detection of biochemical recurrent prostate cancer as previously described. However, the precursors of both 18F-DCFPyL and 18F-PSMA-1007 have proprietary ownership (Progenics Pharmaceuticals and ABX, respectively) and have limited availability. Therefore, it would be advantageous to have other economically viable alternatives of 18F-labeled PSMA tracers for PET imaging.
The labeling of biomolecules such as peptides through Al[18F]-chelation is a new approach that was originally patented by Immunomedics, Inc. 100 and has recently received much attention. In this method, 18F− is first bound to Al3+ to form an aluminum fluoride moiety and then chelated by a suitable chelator. When the chelator is attached to a molecule of interest, radiolabeling the molecule with 18F becomes possible. 101 As the hexadentate ligand N, N-bis(2-hydroxybenzyl) ethylenediamine-N,N-diacetic acid has shown a strong affinity toward Al[18F]-based radiolabeling, due to its fast chelating kinetics at room temperature and favorable stability constant (log K = 24.8) with aluminum, labeling PSMA-11 with 18F through Al[18F] is reliably rapid, usually within 15 min at room temperature. 102,103 This success combines the proven pharmacophore with an ideal PET isotope for PSMA imaging.
According to preclinical evaluations performed by Lütje et al., Al[18F]-PSMA-11 is unstable in water, but has a high internalization rate in PSMA-expressing tumors and lower renal accumulation compared to 68Ga-PSMA-11. 104 A recent clinical study performed in 15 patients at Uruguayan Center of Molecular Imaging showed that a significantly higher SUVmax ratio for Al[18F]-PSMA-11 compared to 68Ga-PSMA-11 was observed (median value: 10.7 vs. 6.0). 105 The data suggest that Al[18F]-PSMA-11 PET/CT is a promising imaging technique for the evaluation of patients with prostate cancer. To facilitate the clinical application of Al[18F]-PSMA-11, the authors have recently developed the fully automated procedure as a simple single-step one-pot reaction in a modified TRACERlab FXN synthesis module (GE Healthcare, Chicago, IL). Al[18F]PSMA-11 can be reliably produced at the range of 1.11–1.85 GBq (300–500 mCi; ≥95% radiochemical purity) in their current setting, with up to 60% radiochemical yield (nondecayed corrected) and the total synthetic time <30 min. In addition, the authors have successfully overcome the stability issue using 10 mM phosphate-buffered saline with 8% EtOH as the formulated solution, in which Al[18F]PSMA-11 remains stable during the observation process at room temperature.
In addition to Al[18F]PSMA-11, Al[18F]PSMA-BCH has also been recently developed and showed excellent detectability of tumor lesions. 106 All of these indicate that Al[18F] can be a convenient labeling strategy that has a great potential to accelerate the preparation of 18F radiopharmaceuticals in PSMA-targeted imaging.
Develop J591 antibody fragments to optimize pharmacokinetics
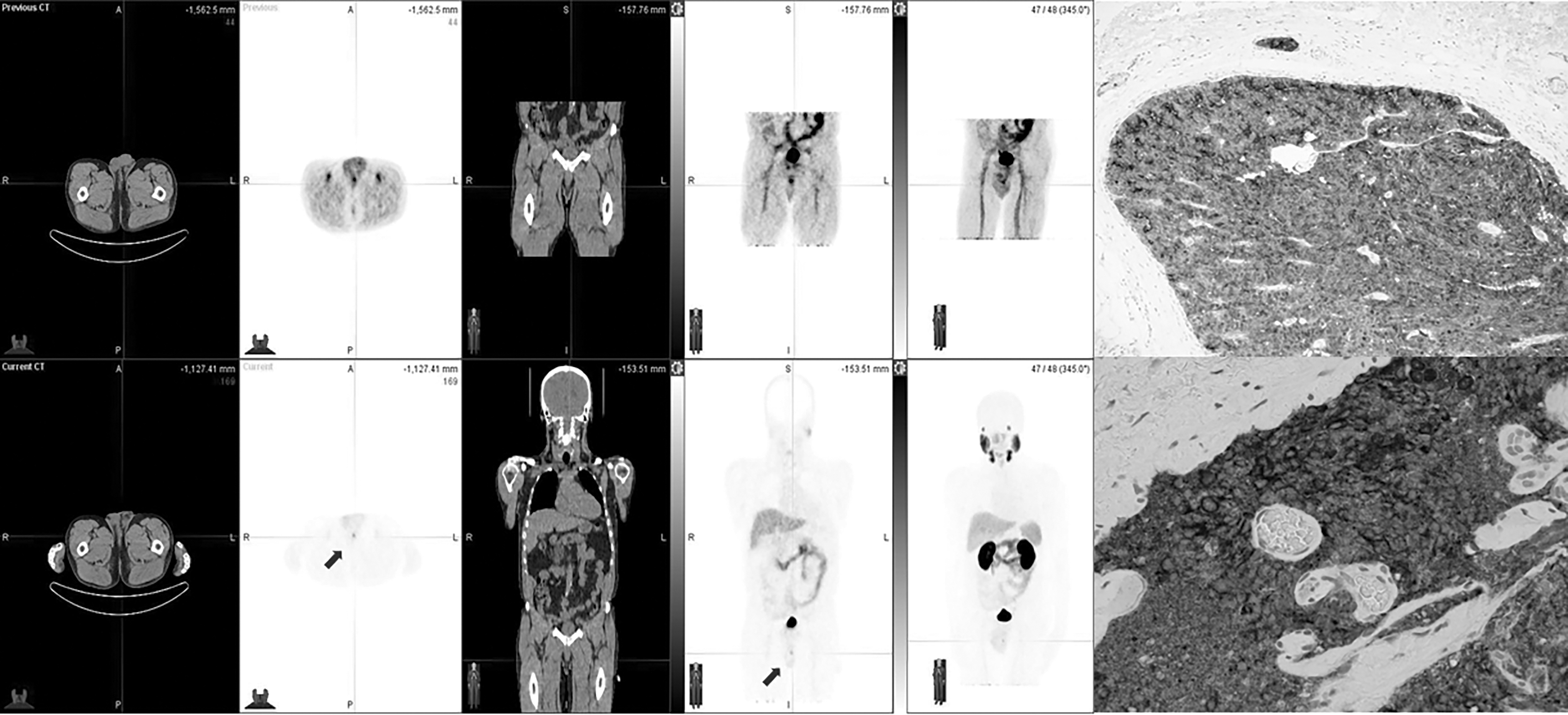
As previously discussed, J591 has great targeting capability toward PSMA but suffers from slow tumor uptake in vivo. However, compared to PSMA inhibitor-based radiopharmaceuticals, the accumulation of J591 in salivary gland is significantly lower for minimizing the occurrence of xerostomia. Therefore, studies have been focused on downsizing J591 to improve its pharmacokinetics. 107 –111 One of the most promising studies using this concept is an anti-PSMA minibody, 89Zr-Df-IAB2M, which has been recently evaluated in 15 patients and demonstrated favorable biodistribution and kinetics for targeting metastatic prostate cancer (Fig. 8). 108 Further investigations may reveal the potential of IAB2M in PSMA TAT when the molecule is labeled with alpha-emitters such as 212Pb and 225Ac.
Lesion targeting with 89Zr-IAB2M in mPC patient.
Utilize albumin binding moieties to enhance overall tumor uptake
Plasma protein binding can be an effective strategy to improve the pharmacokinetic properties in decreasing clearance rate, as well as enhancing specific uptake of drugs. 112 As a result, several investigations have been focused on optimizing the biodistribution of low molecular weight PSMA ligands through the introduction of an albumin binding moiety. 113 –116 A noteworthy process is conjugating Evans Blue, a strong albumin binding moiety, with a therapeutic PSMA-inhibitor, which has been currently investigated by several groups. 117 –119 In a first proof-of-concept study, 177Lu-PSMA-617 conjugated with Evans Blue (177Lu-EB-PSMA-617) was evaluated in 9 patients with metastatic castration-resistant prostate cancer. The tumor uptake was about 2.15-fold higher than that in patients receiving 177Lu-PSMA-617 (Fig. 9). 119 Although higher uptakes in kidneys and red bone marrow of 177Lu-EB-PSMA-617 are also observed compared to 177Lu-PSMA-617, the treatment was well tolerated in this pilot study at low doses. Further clinical studies with increasing therapeutic dose and frequency of 177Lu-EB-PSMA-617 administration will be needed to fully investigate the benefit of utilizing albumin binding moieties in PSMA ligands.

Radiohybrid PSMA inhibitors as a new generation of theranostic agents
With the aim to efficiently label peptide-based radiopharmaceuticals such as PSMA inhibitors with either 18F or radiometals, a new generation of radiopharmaceuticals named radhybrid PSMA (rhPSMA) ligands has recently been developed by Wurzer et al. 120 A unique feature of such rhPSMA ligands is that both fluorine and the metal are always present, but only one of them as radioactive isotope (e.g., [18F,natLu]rhPSMA or [ 19 F,177Lu]rhPSMA). As chemically identical twins, rhPSMA could be used to bridge imaging and therapy. For example, information regarding dosimetry and/or therapeutic monitoring can be acquired from [18F,natLu]rhPSMA imaging, while [ 19 F,177Lu]rhPSMA can be used for corresponding PSMA RLT. Recent report of 18F-rhPSMA-7 demonstrates its strong potential in imaging patients with prostate cancer. 121 It has currently been licensed by Blue Earth Diagnostics for further clinical development. 122
Conclusions
Over the last decade, the development of radiopharmaceuticals for PSMA-targeted imaging and therapy has become one of the most active and dynamic fields of research in nuclear medicine. According to Chang et al., 80.2% of prostate malignant cells overexpress PSMA, and the expression of PSMA correlates well with serum PSA concentration. 5 Therefore, there is little doubt that PSMA will remain an important focus of research in both academia and industrial companies. At this stage, there are several ongoing clinical trials of PSMA-targeted radiopharmaceuticals. Results of prospective phase III clinical trials will soon be available to reveal the diagnostic/therapeutic benefits on the predetermined endpoints of these agents.
Authors' Contributions
Each named author has substantially contributed to drafting this article. M.L.: Draft the outline and determine the subtopics of this article; lead the research of 68Ga production and Al[18F] radiolabeling in the facility. R.T.T.: Summarize the development of radiopharmaceuticals in PSMA-targeted imaging and therapy; provide their recent progress in 68Ga production through a medical cyclotron and the synthesis of Al[18F]PSMA-11. K.K.: Provide the detailed description and clinical data regarding 68Ga-PSMA-11, 177Lu-PSMA-617, and 177Lu-PSMA I&T. D.B.L.: Assist data interpretation and article preparation. G.C.R.: Supervise the data collection and article preparation. The article has been read and approved for submission by all the named authors.
Footnotes
Disclosure Statement
Dr. Gregory C. Ravizzini is the recipient of research funding from Blue Earth Diagnostics and Advanced Accelerator Applications.
Funding Information
This work is supported by the CABI reinvestment fund through the University of Texas MD Anderson Cancer Center.