
Abstract
Background:
Colorectal cancer (CRC) is a significant public problem and the third cause of cancer-induced death all over the world. Long noncoding RNA (lncRNA) has been reported as a vital mediator in human cancer. However, the precise role of lncRNA myocardial infarction associated transcript (MIAT) in CRC is unclear.
Materials and Methods:
The abundance of MIAT, miR-488-3p, and the type 1 insulin-like growth factor receptor (IGF1R) was measured by real-time quantitative polymerase chain reaction assay. Western blot assay was carried out to assess the protein level in CRC samples or control group. The cell activity, abilities of migration and invasion, and glycolysis were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazol-3-ium bromide (MTT), transwell, and testing glucose consumption and lactate product, correspondingly. The target association between miR-488-3p, MIAT, or IGF1R was predicted and established by bioinformatics tools, dual-luciferase reporter, and RNA pull-down assays, correspondingly. The effects of MIAT silencing in vivo were analyzed by animal experiments.
Results:
LncRNA MIAT was upregulated in CRC sample and that was positively correlated with IGF1R expression. Loss-of-functional assay suggested that knockdown of MIAT impeded cell activity, migration, invasion, and glycolysis of CRC cells in vivo, along with xenograft growth in vivo. Moreover, silencing of IGF1R inhibited the progression of CRC. Therefore, overexpression of IGF1R could abolish silencing of MIAT-induced effects on CRC cells. Mechanistically, MIAT was a sponge for miR-488-3p, thereby regulating IGF1R expression in CRC.
Conclusion:
The present study confirmed that the “MIAT/miR-488-3p/IGF1R” pathway was involved in the development of CRC, which may be the target for developing therapeutic approaches for CRC.
Introduction
Colorectal cancer (CRC) is the third primary cause of cancer death all over the world. 1 Despite important development made recently in the judgment and management of CRC, the outcomes of CRC were disappointing due to tumor recurrence and metastasis. 2,3 Consequently, an extensive investigation of the underlying mechanisms of CRC process might help the development of new therapeutic strategies for CRC, which is urgently needed to prolong patient survival.
Long noncoding RNAs (lncRNAs; length >200 nucleotides) occupy 80% of the proportion in all noncoding RNAs. 4 Increasingly abnormal expression of lncRNA was found to take part in the origination and process of CRC. For instance, Wang et al. reported that lncRNA TTN-AS1 could function as a possible treatment target for CRC patients. 5 Furthermore, evidence is increasingly supporting that dysregulation of lncRNA was closely associated with various biological processes, including proliferation, 6 metastasis, 7 inflammation, 8 and angiogenesis. 9 According to predecessor's report, upregulation of myocardial infarction associated transcript (MIAT; accession number: NR_003491) can be exploited as new biomarker and therapeutic target for neuroendocrine prostate and gastric cancer. 10,11 A recent study has revealed that MIAT was also aberrantly expressed in CRC and induced tumor initiation through promoting growth and metastasis of CRC cells, suggesting the oncogenic feature of MIAT in malignancy. 12
MiRNA, a class of small (19–25 nucleotides) noncoding RNA, could act as posttranscriptional gene controllers. 13 Many recent studies demonstrated that dysregulation of tumor-related miRNA played a critical function in CRC development and progression. Masuda et al. reported that miR-488-3p (accession number: NM_030163) level was highly related to recurrence-free survival of breast cancer patients, 14 suggesting that miR-488-3p could serve as independent biomarkers for breast cancer. Moreover, the oncogenic role of Plant homeo domain finger protein 8 (PHF8) in human CRC was associated with its interaction with miR-488. 15 MiR-488 was involved in epithelial–mesenchymal transition of CRC cells through the MAPK signaling pathway by targeting claudin-2. 16
In addition, a large body of evidence has indicated that insulin-like growth factor (IGF) signaling axis was involved in the therapy resistance and tumorigenesis, 17 as well as aerobic glycolysis. 18 The type 1 insulin-like growth factor receptor (IGF1R; accession number: NM_000875) was a key membrane receptor for IGF-I. Evidence has indicated that IGF1R activation can regulate mitogenesis, angiogenesis, transformation, apoptosis, and cell motility thereby serving a key function in carcinogenesis. 19 In addition, activated IGF1R could phosphorylate several substrates, thereby recognizing phosphatidylinositol 3-kinase, growth factor receptor-bound 2 (GRB2), and SH2-containing protein tyrosine phosphatase 2 (SHP2/Syp), which led to activation of downstream signaling pathways such as MAPK and Phosphatidylinositide 3-kinases/protein kinase B signaling pathway. 20
Therefore, in this article, the expression level and carcinogenic role of MIAT in CRC were explored. The regulator mechanism of MIAT in CRC was probed by performing function experiments and rescue assays. In summary, the authors aimed to discover the function and regulatory pattern of MIAT/miR-488-3p/IGF1R in CRC development.
Materials and Methods
Patient specimens
Tumor tissues and adjacent nontumorous samples (located >3 cm from the CRC tissues) were harvested surgically from 30 patients with CRC at the First People's Hospital of Tianmen. None of the patients received radiotherapy, chemotherapy, or other therapies before surgical operation. Samples were stored at a −80°C refrigerator before further analysis. The experimental procedures were approved by the Ethics Committee of First People's Hospital of Tianmen. In addition, written informed consent was offered by all patients according to the principles outlined in The Declaration of Helsinki. The clinicopathologic features of these patients were shown in Table 1.
The Clinicopathologic Features of Colorectal Cancer Patients
Cell culture
The human colon mucosal epithelial cell line (NCM460) was gained from the Chinese Academy of Sciences (Shanghai, China). CRC cell lines (LOVO; ATCC® CCL-229™, SW480; ATCC® CCL-228™, SW620; ATCC® CCL-227™, and HT29; ATCC® HTB-38™) were acquired from the American Type Culture Collection (Rockville, MD). The above cells were maintained in RPMI 1640 medium (Wisent, Shanghai, China) supplemented with 10% fetal bovine serum (Biochrom KG, Berlin, Germany) and 100 U/mL of penicillin and 100 U/mL of streptomycin (Solarbio, Beijing, China) in an incubator with 5% CO2 at 37°C.
Real-time quantitative polymerase chain reaction
Total RNA was isolated using TRIzol reagent (Takara, Dalian, China) following its recommendations. The concentration of RNA was assessed on NanoDrop 2000c (Thermo Fisher Scientific, Carlsbad, CA). RNA was reverse transcribed to complementary DNA with PrimeScript™ RT Reagent Kit (Takara) and Mir-X miR First-Strand Synthesis Kit (Takara), respectively. The real-time quantitative polymerase chain reaction (RT-qPCR) was performed on AB7300 thermo-recycler (Applied Biosystems, Foster City, CA) using SYBR Select Master Mix (Thermo Fisher Scientific) with specific primers as described by Liu et al. 21 The transcript levels of MIAT, miR-488-3p, and IGF1R were evaluated based on the 2−ΔΔCt method and were standardized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or endogenous small nuclear RNA U6.
The sequences of primers were shown as follows:
MIAT (up, 5′-CTTCCAGAGTCATCCTGTGG-3′; down, 5′-CCAAGGGATCCCCAAGGCTC-3′ Amplicon Size: 239 bp);
miR-488-3p (up, 5′-GCCGAGTTGAAAGGCTAT-3′; down, 5′-CTCAACTGGTGTCGTGGA-3′ Amplicon Size: 69 bp);
IGF1R (up, 5′-TCGACATCCGCAACGACTATC-3′; down, 5′-CCAGGGCGTAGTTGTAGAAGAG-3′ Amplicon Size: 243bp);
GAPDH (up, 5′-TCCCATCACCATCTTCCAGG-3′; down, 5′-GATGACCCTTTTGGCTCCC-3′ Amplicon Size: 145 bp);
U6 (up, 5′-AACGAGACGACGACAGAC-3′; down, 5′- GCAAATTCGTGAAGCGTTCCATA-3′ Amplicon Size: 272 bp).
Western blot assay
CRC cells and tissue were collected and washed with 1 × phosphate buffered saline and lysed in Radio-Immunoprecipitation assay (RIPA; Thermo Fisher Scientific) buffer. Immunoblotting was performed using standard procedures and various antibodies. Briefly, proteins in the supernatant were extracted by high-speed centrifugation and quantified using bicinchoninic acid (BCA) protein assay (Bio-Rad, Hercules, CA). Thirty micrograms of protein for each sample was subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then electroblotted onto polyvinylidene fluoride membranes (Thermo Fisher Scientific). Then, the membranes were enshrouded with 3% Albumin Bovine V (Amyjet Scientific, Wuhan, China) for 2 h. The antibodies were added into the membranes and incubated at 4°C overnight with GAPDH (ab181602; 1:3000 dilution; Abcam, Cambridge, MA) as control, followed by goat anti-rabbit IgG-H&L (#7074; 1:3000 dilution; Cell Signaling Technology, Danvers, MA). Finally, Western band was visualized and analyzed with ECL Western Blotting Detection Kit (Solarbio) and Image Lab software 5.2 (Bio-Rad), respectively. The antibodies were listed as follows: IGF1R (ab182408; 1:1000 dilution; Abcam), glucose transport protein type 1 (GLUT1; ab115730; 1:1000 dilution; Abcam), and Lactate dehydrogenase (LDHA; ab101562; 1:1000 dilution; Abcam).
Transfection assay
Specific short hairpin RNA (shRNA) targeting MIAT (sh-MIAT), or IGF1R (sh-IGF1R) and their scrambled control (sh-NC), miR-488-3p mimic (miR-488-3p) and its control (miR-NC), and miR-488-3p inhibitor (anti-miR-488-3p) and its control (anti-NC) were purchased from RiboBio (Guangzhou, China). The transfection mixture (100 nM) was dissolved in serum-free medium and added to the cells (2.5 × 105). In addition, IGF1R-overexpressing pcDNA-plasmid (pcDNA-IGF1R; 100 nM; designed by RiboBio) or its negative control (pcDNA) was transfected into CRC cells using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) in compliance with the user's guidebook. After 48 h, the cells were collected for protein/RNA extraction and other experiments.
Cell proliferation assay
Three-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazol-3-ium bromide (MTT) analysis was carried out to assess the viability of CRC cells. Approximately 1 × 103 of LOVO and SW480 cells were cultivated into a 96-well plate. After incubation in the medium at 37°C for specified time, 20 μL MTT reagent (Beyotime, Shanghai, China) was added to the cells and allowed to react for 4 h. After that, the dimethyl sulfoxide was added to 96-well plate and incubated for 10 min. The cell proliferation curves were plotted by testing absorbance (490 nm) on a microplate reader (Applied Biosystems).
Transwell assay
Cell migration capability was assessed by transwell migration assay with 24-well transwell chamber that had 8 μm pore size membranes. CRC cells (2 × 105 cells) were placed in the upper chamber with 200 μL of serum-free medium, while complete medium in bottom chamber was used to induce cell migration. Following incubation at 37°C for 24 h, the cells on the basal side of the membrane were fixed and then dyed with methanol and 0.1% crystal violet. Each well was imaged under a microscope (Leica, Wetzlar, Germany). In addition, for invasion assay, upper chambers were covered with Matrigel (BD Biosciences, San Jose, CA), and the other experimental procedures were unchanged.
Measurement of glucose uptake and lactate product
Glucose Assay Kit (Solarbio) and Lactate Assay Kit (Solarbio) were utilized to analyze glycolysis ability in CRC cells. Briefly, 1 × 105 of LOVO and SW480 cells per well were sowed in 24-well plates. After transfection 48 h, the culture medium supernatant was collected, and cells were homogenized and centrifuged to test glucose concentration and lactate product referring to their recommendations, individually.
Dual-luciferase reporter assay
The bioinformatics tool (
RNA pull-down assay
LOVO and SW480 cells were introduced with bio-miR448-3p with biotin labeled or control (bio-miR-NC). After 48 h, CRC cells were collected and then lysed in RIPA buffer, and then 100 μL of cell lysates were incubated with streptavidin magnetic beads (Invitrogen) to pull down the target RNA at 4°C for 4 h. After being washed with wash buffer, RNA complexes bound to the beads RNA–RNA complex were centrifuged and purified before RT-qPCR assay.
Xenografted animals
The animal experiments were approved by the Ethics Committee of the First People's Hospital of Tianmen. BALB/c nude mice, 4–6 weeks old, were purchased from Model Animal Research Center at Nanjing University (Nanjing, China). One × 106 LOVO cells (stably infected with sh-MIAT or sh-NC) were hypodermically vaccinated into the left axilla of nude mice (n = 6). Tumor growth was monitored every week, and volume was calculated using the formula: V = 1/2 × ab 2 method [maximum (a) and minimum (b) length of the tumor]. After 5 weeks, the mice were sacrificed, and the tumors were dissected for further analysis.
Statistical analyses
All data were exhibited as mean ± standard deviation. Differences in two treatment groups or multiple groups were assessed by Student's t-test or one-way analysis of variance, respectively, and p < 0.05 was regarded as statistically significant. GraphPad Prism 7 (GraphPad, La Jolla, CA) was used for statistical analyses.
Results
MIAT and IGF1R were upregulated in CRC tissues
Through RT-qPCR and Western blot analyses within CRC tumor and neighboring nontumor tissues, the authors observed that MIAT expression was notably strengthened in CRC samples compared with the negative control (Fig. 1A). In addition, IGF1R mRNA was overexpressed in CRC tissues in comparison with nontumor tissues; consistently, the authors also found the increase of protein expression level of IGF1R in CRC samples (Fig. 1B, C). Importantly, IGF1R was positively correlated with MIAT in CRC samples (Fig. 1D). Therefore, these results implied that the upregulation of MIAT and IGF1R was correlated with CRC development.
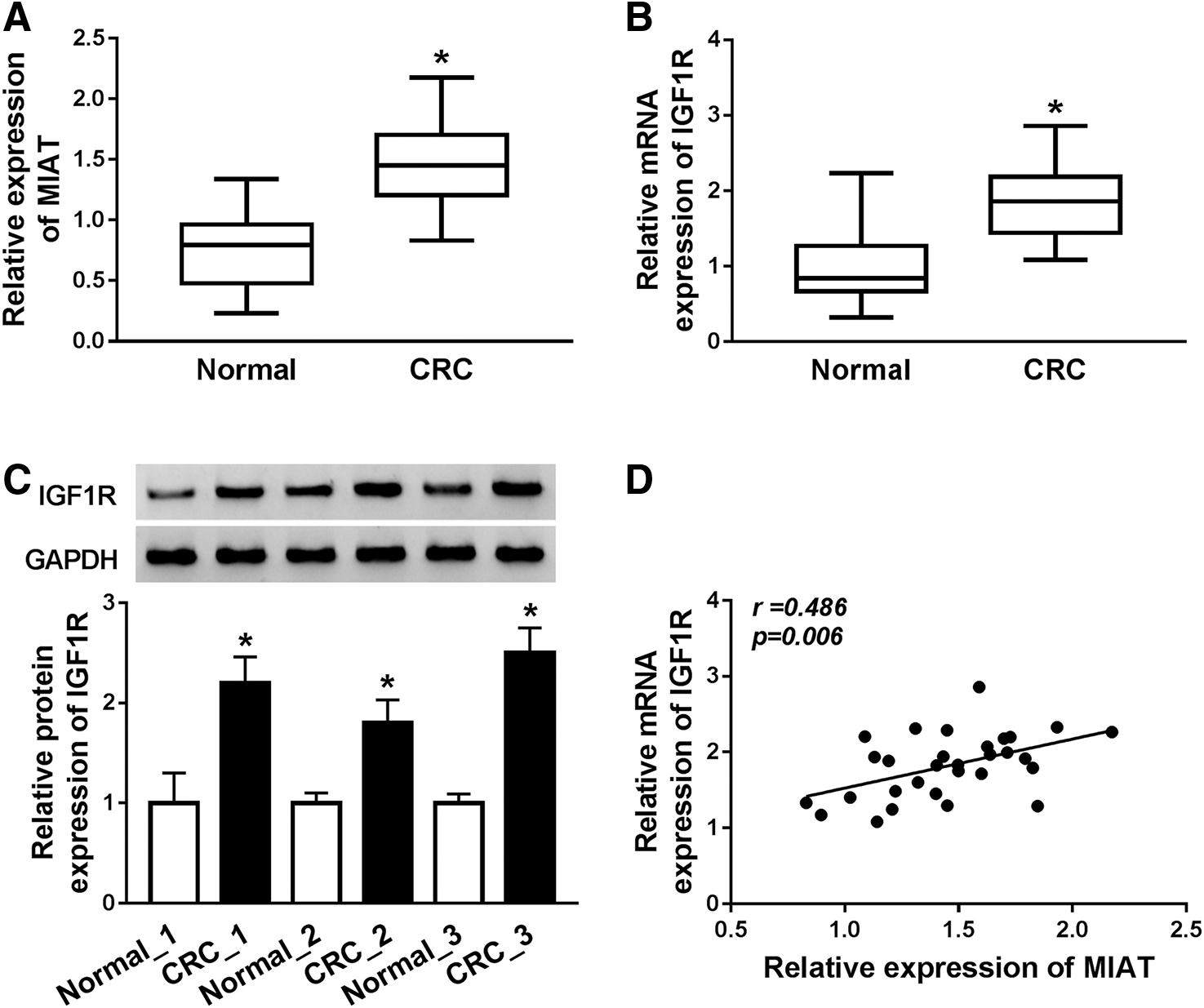
The expression levels of MIAT and IGF1R in colorectal cancer tissues.
Knockdown of MIAT impeded proliferation, migration, invasion, and glycolysis in CRC cells
Initially, the authors found that MIAT was reduced in LOVO and SW480 cells with respect to NCM460 cells (Fig. 2A). Furthermore, sh-RNA against MIAT was designed to knock down MIAT level in LOVO and SW480 cells, and MIAT was reduced more than half in CRC cells than control group, suggesting that knockdown experiments were successful (Fig. 2B). MTT assay revealed that silencing of MIAT obviously reduced cell proliferation in LOVO and SW480 cells (Fig. 2C). Moreover, transwell migration and invasion analyses revealed that silence of MIAT obviously hindered the cell mobility, including migration and invasion (Fig. 2D, E). For glycolysis assay, compared with sh-NC group, a remarkable decrease in glucose consumption and lactate secretion was found in CRC cells after sh-MIAT transfection (Fig. 2F, G). In addition, GLUT1 and LDHA were detected by Western blot analysis. Downregulation of MIAT triggered a decrease of GLUT1 and LDHA in LOVO and SW480 cells than sh-NC group (Fig. 2H, I). Collectively, MIAT silencing impaired development of CRC.
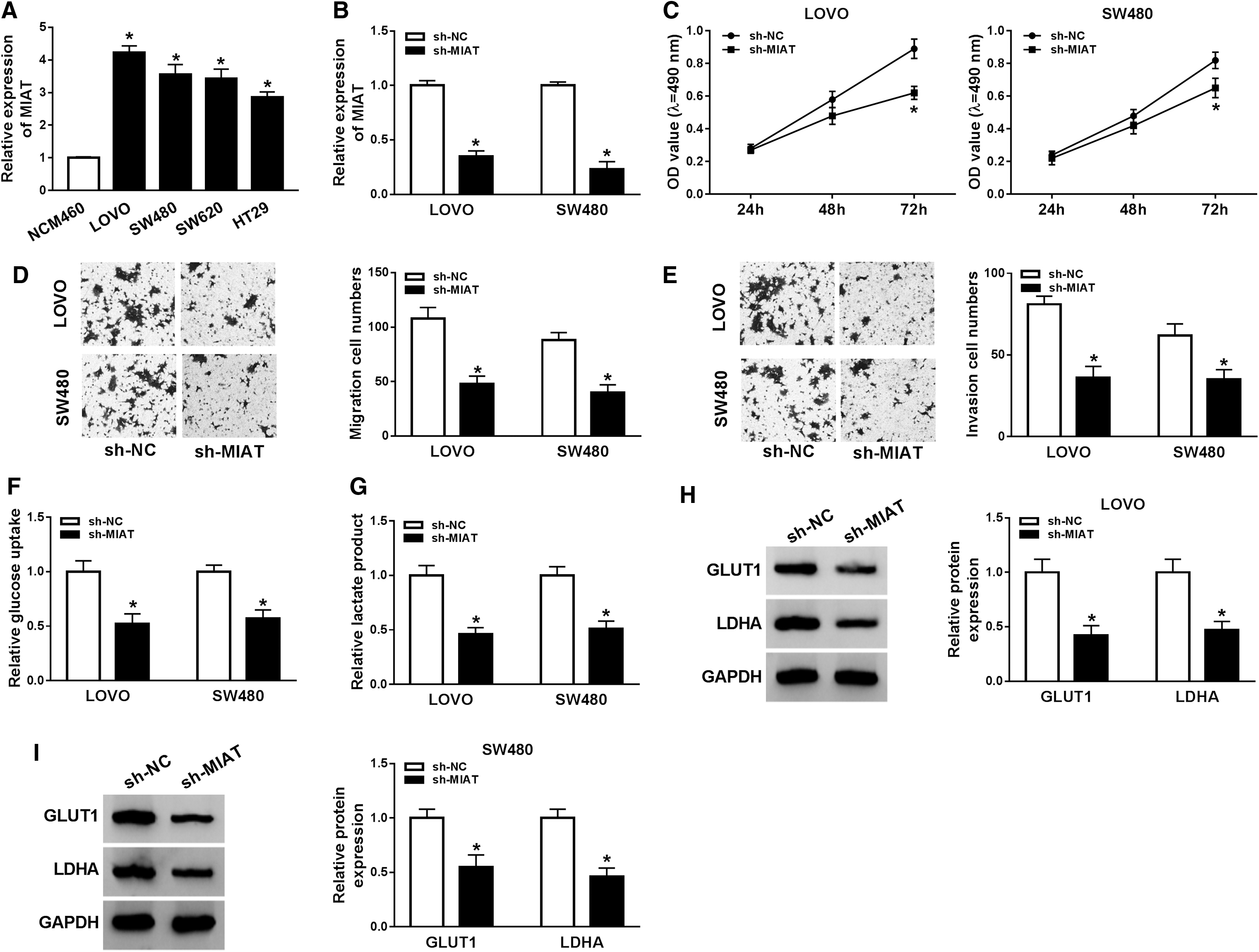
Effects of MIAT silencing on colorectal cancer cell proliferation, migration, invasion, and glycolysis.
Downregulation of IGF1R restrained CRC cell proliferation, migration, invasion, and glycolysis
Functional experiments were performed to probe the role of IGF1R in CRC progression. Based on that, IGF1R was upregulated in CRC cells by Western blot analysis, as indicated in Figure 3A. Afterward, the authors transfected the sh-IGF1R into LOVO and SW480 cells and found that protein expression level of IGF1R was dramatically dropped in sh-IGF1R group than that in sh-NC group (Fig. 3B). Downregulation of expression of IGF1R led to a notable downregulation in cell proliferation in both LOVO and SW480 cells (Fig. 3C). In addition, migration and invasion capabilities of CRC cells were repressed by knockdown of IGF1R (Fig. 3D, E). Silencing of IGF1R decreased glucose consumption and lactate production in LOVO and SW480 cells than control group (Fig. 3F, G). The authors also observed that GLUT1 and LDHA were declined after IGF1R knockdown in LOVO and SW480 cells by performing Western blot analysis (Fig. 3H, I). The above results revealed that IGF1R regulated proliferation, migration, invasion, and glycolysis to participate in CRC progression.
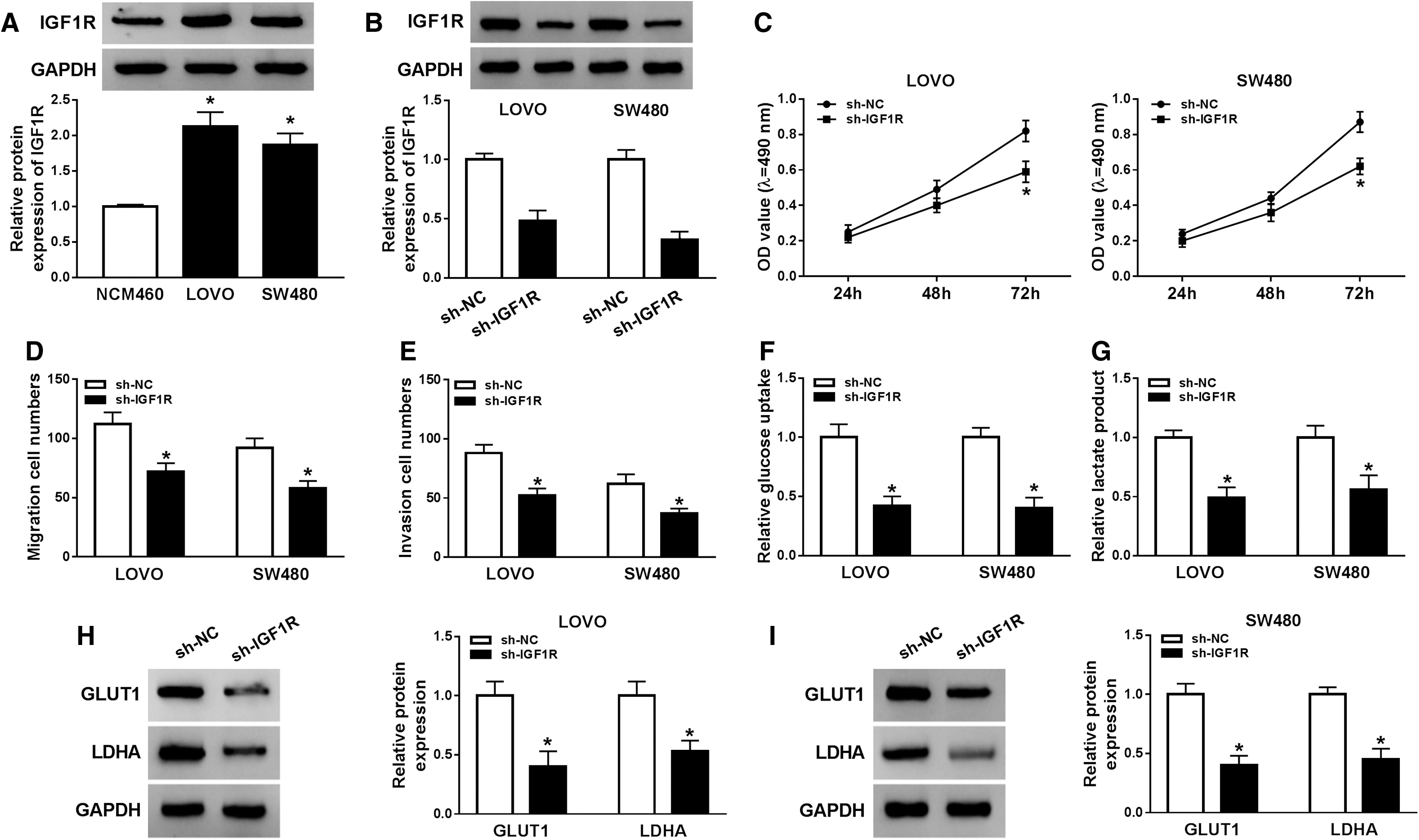
The influence knockdown of IGF1R on colorectal cancer cell proliferation, migration, invasion, and glycolysis.
Overexpression of IGF1R overturned MIAT silence-mediated inhibitory effects on proliferation, migration, invasion, and glycolysis
The association between IGF1R and MIAT was further studied, and the authors found that IGF1R expression decreased in LOVO and SW480 cells after infection with sh-MIAT while increased after IGF1R overexpression, as demonstrated by Western blot assay (Fig. 4A, B). Furthermore, pcDNA-IGF1R overturned the suppressive role of MIAT silencing on cell activity in LOVO and SW480 cells (Fig. 4C, D). More importantly, migration and invasion were restrained in LOVO and SW480 cells after MIAT was knocked down, while MIAT silence-mediated suppression effects were partly rescued by overexpression of IGF1R (Fig. 4E, F). The authors also found that after transfection with pcDNA-IGF1R into LOVO and SW480 cells, sh-MIAT silence-mediated suppressing effects on glycolysis by decreasing glucose consumption and lactate production, along with GLUT1 and LDHA expression, were partly counteracted (Fig. 4G–J). In summary, all evidence implied that IGF1R was a significant regulator of MIAT mediated proliferation, migration, invasion, and glycolysis in CRC.
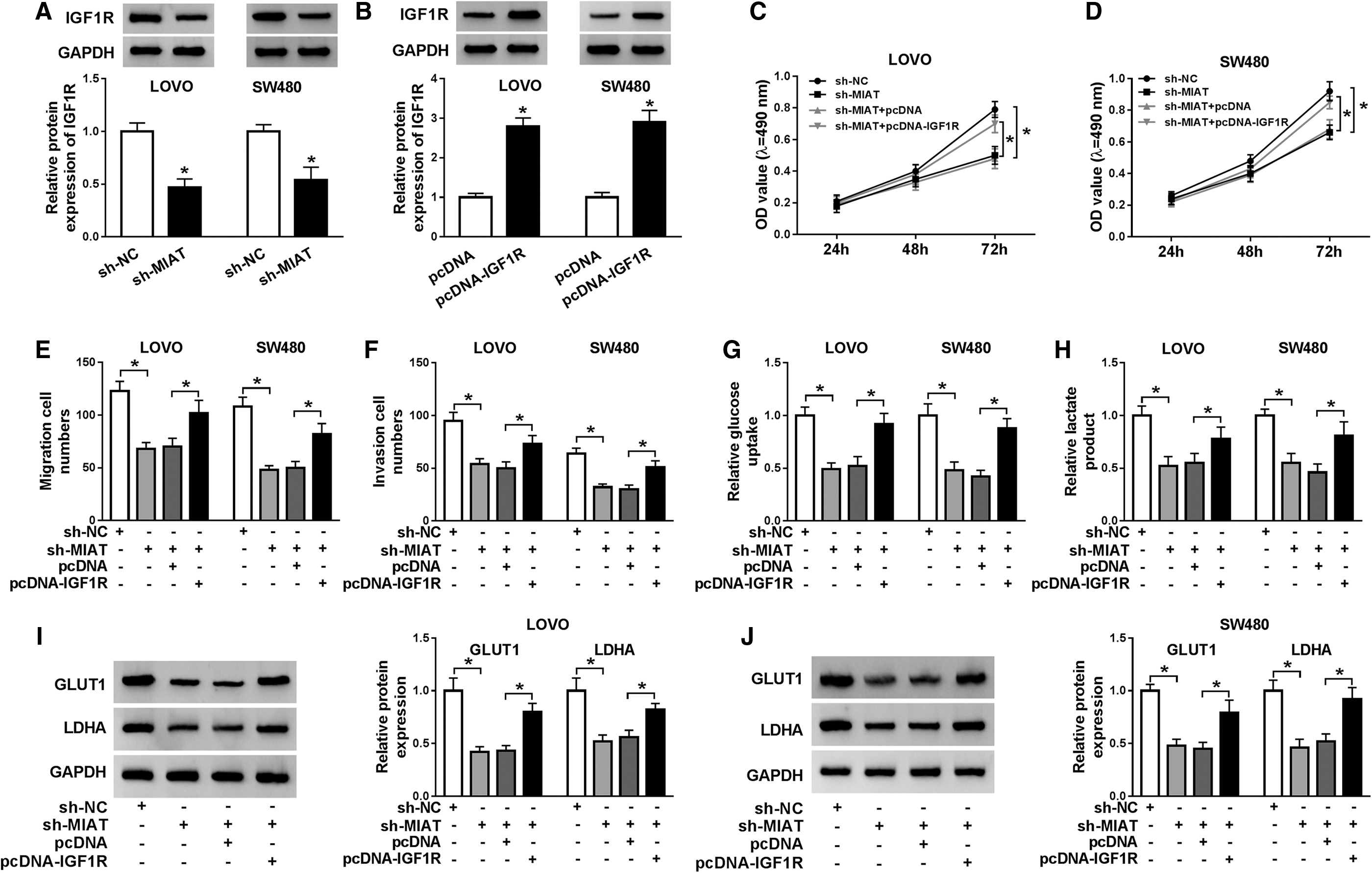
Overexpression of IGF1R could abolish silencing of MIAT-mediated effects on proliferation, migration, invasion, and glycolysis.
MIAT regulated IGF1R by targeting miR-488-3p in CRC
Binding sites between miR-488-3p and MIAT, as well as matched mutated nucleotides, are presented in Figure 5A. Analogously, the results of starBase assay also indicated that miR-488-3p had binding sites in 3′UTR of IGF1R (Fig. 5B). Then, the authors examined the luciferase activity in LOVO and SW480 cells, as shown in Figure 5C and D, and luciferase activity of wild type-MIAT drastically decreased in miR-488-3p group; besides, mutated-MIAT was not affected by miR-488-3p than control group (Fig. 5C, D). Similar results were found in Figure 5E and F, suggesting that miR-488-3p could bind to 3′UTR of IGF1R. Furthermore, the results of RNA pull down revealed that MIAT and IGF1R expression were remarkably higher in bio-miR-448-3p group than control group (Fig. 5G, H). MiR-488-3p was overexpressed in LOVO and SW480 cells transfected with sh-MIAT (Fig. 5I). In addition, transfection with miR-488-3p into LOVO and SW480 cells caused high expression of miR-488-3p, but opposite results were noticed in LOVO and SW480 cells infected with anti-miR-488-3p (Fig. 5J). In addition, IGF1R expression was downregulated in LOVO and SW480 cells after miR-488-3p was overexpressed; importantly, downregulation of MIAT-induced inhibition effect on IGF1R expression was offsetted by downregulation of miR-488-3p (Fig. 5K, L). Consequently, miR-488-3p, interacted with IGF1R, was a target gene of MIAT in CRC. Additional, RIP assay confirmed the association among MIAT, miR-488-3p, and IGF1R in CRC cells (Supplementary Fig. S1).
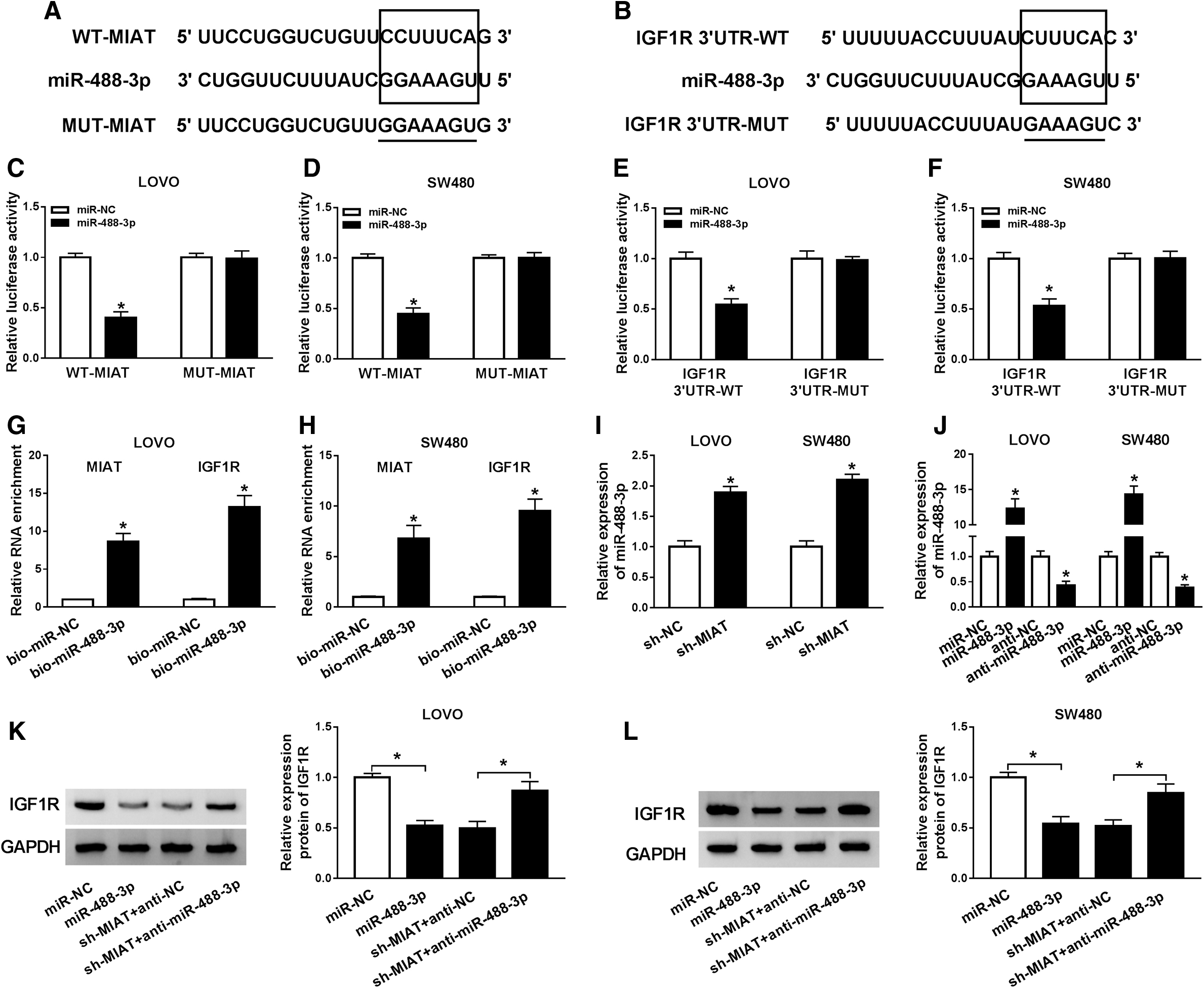
MIAT acted as a ceRNA by sponging miR-488-3p and regulated IGF1R expression.
MIAT inhibition restrained the growth of CRC cells in vivo
Results of in vivo growth of CRC showed that sh-MIAT significantly decreased tumor volume and weight in mice inoculated with MIAT-silenced CRC cells than those inoculated with sh-NC-transfected CRC cells (Fig. 6A, B). Besides, knockdown of MIAT significantly inhibited the lactate production compared with sh-NC-transfected group (Fig. 6C). Consistently, the abundance of MIAT was declined, whereas the abundance of miR-488-3p was increased in MIAT-silenced tumors than sh-NC transfected group (Fig. 6D, E). The authors also observed that IGF1R, GLUT1, and LDHA were decreased in MIAT-silenced tumors than control tumors (Fig. 6F). Thus, MIAT regulated the growth of CRC cells by affecting miR-488-3p and IGF1R expression, as well as glycolysis.
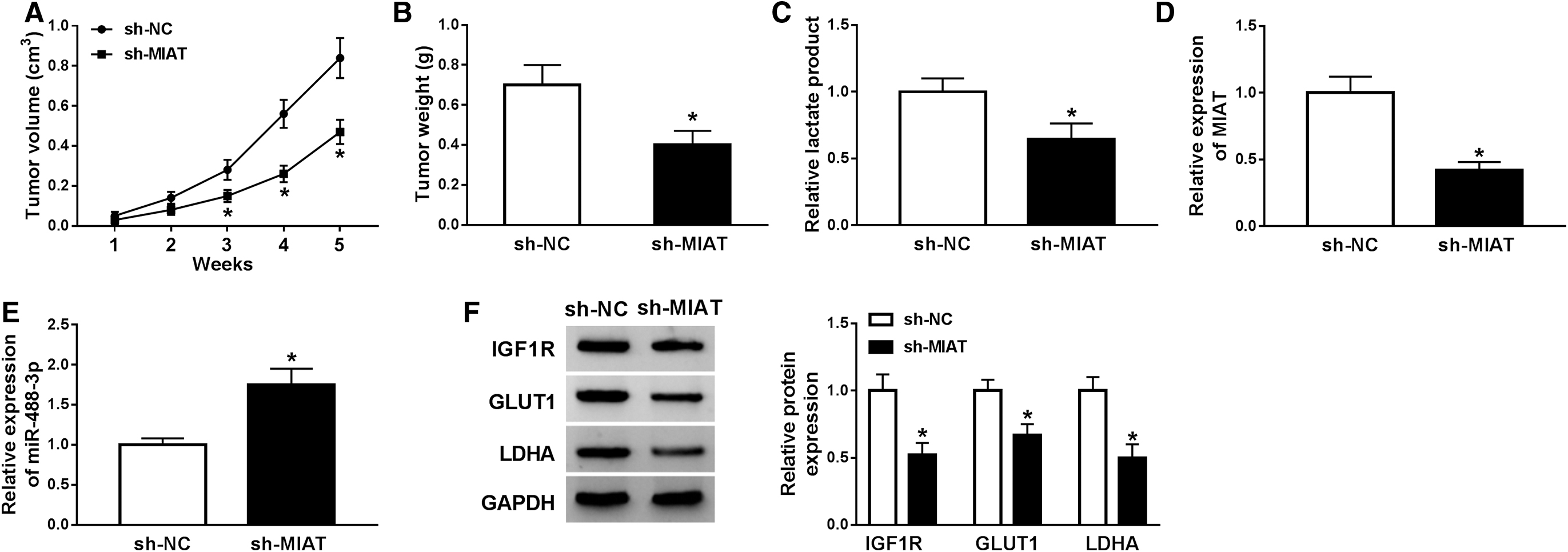
Knockdown of MIAT suppressed the growth of LOVO cells in mice.
Discussion
Currently, MIAT was upregulated in CRC samples than matched negative controls, respectively. The authors also identified the function of MIAT in CRC cells by applying functional approaches. The results demonstrated that silencing of MIAT impeded cell growth, mobility, and glycolysis in CRC cells, and the mechanism of this action sh-MIAT-mediated inhibition effects was related to miR-488-3p/IGF1R pathway, revealing that MIAT functioned as a competing endogenous RNA to upregulate IGF1R by targeting miR-488-3p.
A number of recent articles suggested that MIAT was closely associated with tumor formation and the development; hence, dysregulation of MIAT may also affect the development of malignant tumors. Li et al. demonstrated that MIAT competitively bound to miR-29a-3p to regulate gastric cancer development. 22 Silencing of the MIAT was considered to exert a tumor suppressor effect in melanoma by decreasing cell activity and mobility. 23 In CRC, Liu et al. confirmed that MIAT was an oncogene by promoting proliferation and metastasis of CRC cells. 12 The authors speculated that knockdown of MIAT could impede the CRC process. Based on previous results, MIAT played critical roles in CRC by competitively binding to miRNA to regulate cancer development. Not surprisingly, miR-150-5p, 24 miR-212, 25 and miR-1246 26 were direct target of MIAT in different cancers, more importantly, among which miR-150-5p, miR-212, and miR-1246 were found to play inhibitory roles in the development of CRC. 27 –29 In addition, the interaction relationship between lncRNA and miRNA has attracted attention during the tumorigenic progression. To further investigate the molecular link or crosstalk between MIAT and miR-488-3p, the authors first used bioinformatics analysis and dual-luciferase analysis to validate the binding ability of miR-488-3p on the MIAT. Furthermore, the authors used functional experiments and found that miR-488-3p was negatively regulated by MIAT in CRC cells. In addition, MIAT was more enriched in bio-miR-488-3p than that in control group by performing RNA pull down. To determine the functional role of miR-488-3p in CRC, the authors summarized the possible target gene of miR-488-3p in multiple human cancers. High-mobility group nucleosome binding domain 5 (HMGN5), Paired box 6 (PAX6), and Hairy and enhancer of split-related with YRPW motif protein 2 (HEY2) were direct targets of miR-488 in different cancers, 30 –32 among which PAX6 could induce proliferation and invasion of CRC cell. 33 The authors also found that miR-488 played inhibitory roles in the development of CRC, indicating antioncogenic function of miR-448. Wang et al. reported that miR-488 level was highly related to clinicopathological characteristics of CRC, and overexpression of miR-488 inhibited viability, invasiveness, and metastasis in CRC cells. 16 It is worth mentioning that miR-488-3p could directly bind to the 3′UTR of the 6-phosphofructo-2-kinase/fructose-2, 6-bisphosphatase 3 (PFKFB3), which enhanced glycolysis and lacked phosphatase activity, 34,35 while PFKFB3 is a vital regulator of glycolysis, regulating cell proliferation, vessel aggressiveness, drug resistance, and tumor microenvironment. 36 The authors conjectured that MIAT/miR-488-3p axis could mediate glycolysis ability in CRC cells. Currently, miR-488-3p negatively regulated IGF1R expression. As the authors expected, through a bioinformatics database and dual-luciferase reporter gene assay, IGF1R was verified as a functional target gene of miR-488-3p.
The importance of IGF1R has been well documented in carcinomas. The IGF1R expression level was also upregulated in CRC tumors. 37 Wang et al. reported that downregulation of IGF1R impeded the activity of protein kinase B and limited cellular growth and metabolism. 38 Moreover, downregulation of IGF1R by siRNA in gefitinib-resistant cells could reduce migration and invasion abilities. 39 Their study also confirmed that IGF1R acted as an oncogene and IGF1R was regulated by miR-488-3p in CRC. Considering the regulator relationship among MIAT miR-488-3p and IGF1R, functional experiments were used and confirmed that ectopic overexpression of IGF1R could abolish knockdown of MIAT-induced inhibitory effects on CRC cells.
Collectively, MIAT/miR-488-3p/IGF1R pathway may be connected with development of CRC. By binding to miR-488-3p, MIAT promoted the IGF1R expression, which functioned as a molecular sponge of miR-488-3p.
Conclusion
In summary, the present study suggested that MIAT was upregulated in CRC, and knockdown of MIAT significantly repressed process of CRC. Mechanistically, MIAT acted as a miRNA sponge for miR-488-3p to increase IGF1R expression, which may be a novel target for developing therapeutic strategy to treat CRC.
Footnotes
Disclosure Statement
The authors declare that they have no financial conflicts of interest.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.