
Abstract
Background:
Endometrial cancer (EC) is an intractable gynecological cancer with increasing incidence and mortality worldwide. Accumulating studies indicated that long noncoding RNA nuclear enriched autosomal transcript 1 (NEAT1) was a novel oncogene implicated in a variety of cancers. However, whether NEAT1 could accelerate cell growth in EC is unclear.
Materials and Methods:
NEAT1, microRNA (miR)-202-3p, and T cell immunoglobulin and mucin domain 4 (TIMD4) levels were detected by quantitative real-time polymerase chain reaction. Cell proliferation and apoptosis were examined by cell counting kit-8 and flow cytometry. Transwell assay was employed for the evaluation of cell migration and invasion. The relationship between miR-202-3p and NEAT1 or TIMD4 was determined by luciferase reporter system. TIMD4 protein expression was assessed by Western blot assay.
Results:
NEAT1 was upregulated, whereas miR-202-3p was downregulated in EC tumors and cells. Depletion of NEAT1 restrained EC cell proliferation, migration, invasion, and improved apoptosis. MiR-202-3p was targeted by NEAT1 and could bind to TIMD4. Subsequently, it is observed that miR-202-3p inhibitor neutralized NEAT1 silencing mediated suppression on EC cell progression. Meanwhile, TIMD4 rescued miR-202-3p induced inhibition on cell progression in EC. Furthermore, it was obvious that NEAT1 facilitated TIMD4 expression by absorbing miR-202-3p in EC.
Conclusions:
Upregulation of NEAT1 accelerated EC cell progression through sponging miR-202-3p to facilitate TIMD4 expression, providing potential novel treatment method for EC.
Introduction
Endometrial cancer (EC) is an aggressive cancer and mainly occurred in women, especially young women. 1 It is clarified as EC type I and type II. Type I EC is the common EC with the favorable prognosis. Type II EC with poor prognosis and a high risk of metastasis is relatively high grade and has the histology of serous or clear cell. 2 Although type II accounts for only 10% of EC tumors, it accounts for 40% of all deaths. 3 This study focused on type II EC. The risk factors for EC are complex, such as obesity, estrogen stimulation, infertility, polycystic ovarian syndrome, insulin resistance, and aberrant endometrial hyperplasia. 4,5 The standard treatment for EC is hysterectomy. However, the 5-year survival is still ∼15%–50% due to local recurrence, delayed diagnosis, and distal metastases. 6 –8 Therefore, illumination of the specific pathogenesis genetically might provide effective therapy methods for EC.
Long noncoding RNAs (lncRNAs) are conserved transcripts that regulate cell cycle, metabolism, proliferation, inflammation, metastasis, and apoptosis in multiple diseases. 9 –11 The nuclear enriched autosomal transcript 1 (NEAT1) acted as novel oncogene in various cancers, including melanoma, hepatocellular carcinoma, and cervical cancer. 12 –14 For instance, NEAT1 was overexpressed in retinoblastoma and overexpression of NEAT1 facilitated the development of human retinal pigment epithelium cells by binding to miRNA (miR)-204 and boosting chemokine CXC receptor 4 expression. 15 Similarly, NEAT1 expedited the exacerbation of EC by facilitating cell proliferation, invasion, and inhibiting apoptosis through miR-361/STAT3 axis. 16 NEAT1 sponged miR-23a-3p to upregulate SMC1A expression, thereby accelerating tumorigenesis and progression of acute myeloid leukemia. 17 Thus, the authors suggested that NEAT1 functions as a significant tumor promotor involved in tumorigenesis and progression of EC.
MicroRNAs (miRNAs) are small noncoding RNAs with 16–25 nucleotides and cannot be translated into protein. 18 Increasing data have proved that miRNAs participate in many physiological processes by gene regulation at post-transcriptional level, including messenger RNAs (mRNAs) degradation and protein translation interference. 19 –21 They also participate in cell viability, migration, infiltration, autophagy, apoptosis, and radio resistance in different cancers. 22 For example, miR-202-3p acted as a tumor suppressor in papillary thyroid carcinoma to restrain cell migration and invasion by regulation of wingless (WNT) signaling. 23 Similarly, miR-202-3p could interact with the metastasis-associated lung adenocarcinoma transcript 1/periostin axis to suppress proliferation, epithelial-mesenchymal transition (EMT), and invasion through protein kinase B/mammalian target of rapamycin pathway. 24 Zhao et al. discovered that miR-202-3p was reduced in gastric cancer and played suppressive role in tumor cell development through binding to Gli1. 25 Thus, it is imperative to probe into the effect of miR-202-3p on EC.
The authors purposed to elucidate the biological mechanism of NEAT1 during cell development in EC. Upregulation of NEAT1 in EC tumors and cells suggested the oncogenic role of NEAT1. Silencing of NEAT1 repressed EC cell proliferation, migration, invasion, and accelerated apoptosis, clarified the promotive effects of NEAT1. Significantly, it is discovered that NEAT1 facilitated EC cell development through sponging miR-202-3p to enhance T cell immunoglobulin and mucin domain 4 (TIMD4) expression.
Materials and Methods
Tissue samples
Type II EC patients (n = 20) without preoperative treatment were recruited from Beijing Tongren Hospital. All the EC tumors and the adjacent normal tissues were harvested from those participants by surgery. Those patients have signed informed consent. The protocols were approved by Ethics Committee of Beijing Tongren Hospital.
Cell culture and transfection
EC cells RL95, AN3CA, and neuroendocrine cells (NE) were bought from ATCC (Manassas, VA) and cultured in Dulbecco's modified Eagle's medium (Gibco, Carlsbad, CA) with 0.05% penicillin/streptomycin and 10% fetal bovine serum. Small interfering RNA of NEAT1 (si-NEAT1), control (si-control), cDNA plasmid (pcDNA), and TIMD4 overexpression vector were synthesized from Genepharma (Shanghai, China). MiR-202-3p mimics, miR-202-3p inhibitor (anti-miR-889-3p), and their controls (miR-NC and anti-miR-NC) were purchased from RIBOBIO (Guangzhou, China). Lipofectamine 2000 (Invitrogen, Carlsbad, CA) was employed for cell transfection.
Quantitative real-time polymerase chain reaction
TRIzol reagent (Invitrogen) was applied to isolated total RNA by TRIzol reagent (Invitrogen) was used to synthesize complementary DNA (cDNA) by All-in-One™ First-Strand cDNA. SYBR green (Applied Biosystems, Foster City, CA) was used for quantitative polymerase chain reaction. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 were used to normalize NEAT1, miR-202-3p and TIMD4 levels. The primers were as follows (5′ to 3′): NEAT1 (forward [F], CTTCCTCCCTTTAACTTATCCATTCAC; reverse [R], CTCTTCCTCCACCATTACCAACAATAC); miR-202-3p (F, AGAGGTATAGGGCATGGGAA; R, GGAGACCGCCTGGGAATA); TIMD4 (F, CAAACACGTGCCTTTCACTAACC; R, GACTCAACACTACTCCAGGAATCAC); GAPDH (F, AGGTCGGTGTGAACGGATTTG; R, GGGGTCGTTGATGGCAACA); U6 (F, ACCCTGAGAAATACCCTCACAT; R, GACGACTGAGCCCCTGATG).
Cell counting kit-8 assay
Cell counting kit-8 (CCK-8) assay was applied to determined cell proliferation. In brief, transfected RL95 and AN3CA cells were inoculated on 96-well plates. After proliferation for 24, 48, and 72 h, each well was added with 10 μL CCK-8 (Beyotime, Shanghai, China) and incubated for 2 h. Finally, a microplate reader was used to measure the optical density value at 450 nm.
Flow cytometry
Annexin V-Fluorescein (FITC)/PI apoptosis detection kit (Vazyme, Nanjing, China) was exploited for the analysis of cell apoptosis. In brief, at 48 h after transfection, the cells were collected, centrifuged, resuspended, and stained with Annexin V-FITC and PI for 15 min. Finally, the cells were detected by a flow cytometer.
Transwell assay
Transwell chambers were employed for the detection of migration and invasion. Matrigel was precoated into the upper chamber for invasion assay (without Matrigel for migration assay). Then, transfected RL95 and AN3CA cells were added in the upper chamber for 48 h. Next, 0.1% crystal violet (Sigma, St. Louis, MO) was used to stain the migrated and invaded cells at lower chamber. The migration and invasion rated was measured by a microscope.
Luciferase reporter assay
The fragment of NEAT1 and TIMD4 containing binding sequences of miR-202-3p (NEAT1-WT and TIMD4-WT) and their mutant sequences (NEAT1-MUT and TIMD4-MUT) were constructed into pGL3 luciferase vector. Subsequently, RL95 and AN3CA cells were co-transfected with reporter vector and miR-202-3p or miR-NC for 48 h. Next, luciferase activities were measured through a luminometer (Promega GloMax 20/20 Luminometer).
Western blot
TIMD4 and GAPDH protein were extracted from RL95 and AN3CA cells. Protein expression of TIMD4 and GAPDH was analyzed by Western blot. Polypropylene gel electrophoresis was exploited to separate the proteins. After that, the proteins were transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA) and then blocked by 5% nonfat milk. Primary antibodies against TIMD4 and GAPDH (Abcam, Cambridge, MA) were used to incubate the membranes overnight at 4°C. Finally, the membranes were incubated with secondary antibody (Sangon, Shanghai, China) and detected by an ECL Western blot kit (Beyotime).
Statistical analysis
Data were conducted by SPSS software and GraphPad Prism 7 and showed as means ± standard deviation. Pearson's correlation coefficient was employed to detect the correlation between miR-202-3p and NEAT1 or TIMD4. Statistically significant was determined when p-value <0.05.
Results
Upregulation of NEAT1 in EC
The potential role of NEAT1 during EC development was evaluated by measuring NEAT1 levels in EC tumors and cells using quantitative real-time polymerase chain reaction. As exhibited in Figure 1A, NEAT1 level was evidently elevated in EC tumors relative to the adjacent normal tissues. Meanwhile, NEAT1 was upregulated in EC cell lines (RL95, AN3CA) in comparison with neuroendocrine cells NE (Fig. 1B). Therefore, the authors implicated that NEAT1 might act as oncogene in EC.
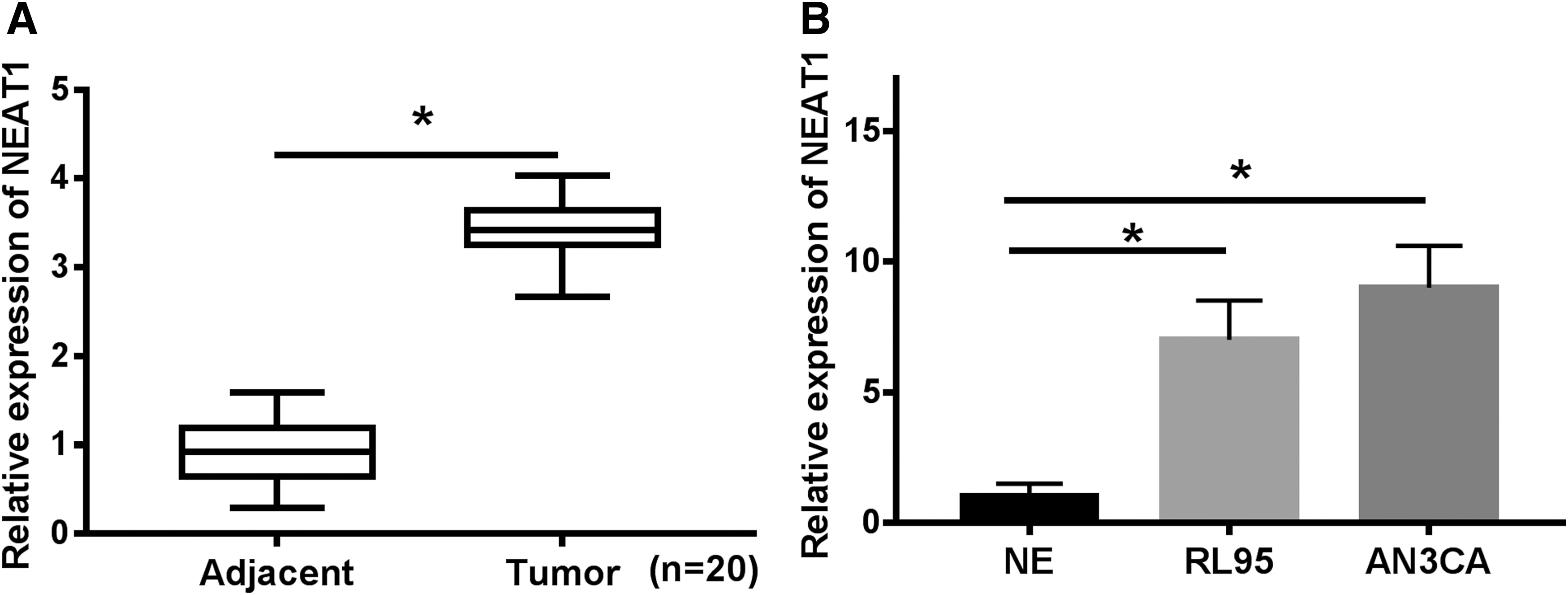
NEAT1 expression was upregulated in EC tumors and cell lines.
Elimination of NEAT1 inhibited cell proliferation, migration, invasion, and facilitated apoptosis in EC
The regulatory effects of NEAT1 on EC cells was investigated by silencing of NEAT1 in EC cells. Apparently, the expression of NEAT1 in RL95 and AN3CA cells was decreased after NEAT1 knockdown (Fig. 2A, B). As expected, proliferation of EC cells was repressed when transfected with si-NEAT1 compared with si-control (Fig. 2C, D). Conversely, cell apoptosis was boosted by NEAT1 silencing (Fig. 2E, F). Synchronously, transwell assay exhibited that depletion of NEAT1 attenuated cell migration and invasion in EC (Fig. 2G, H). Taken together, depletion of NEAT1 inhibited cell progression in EC.
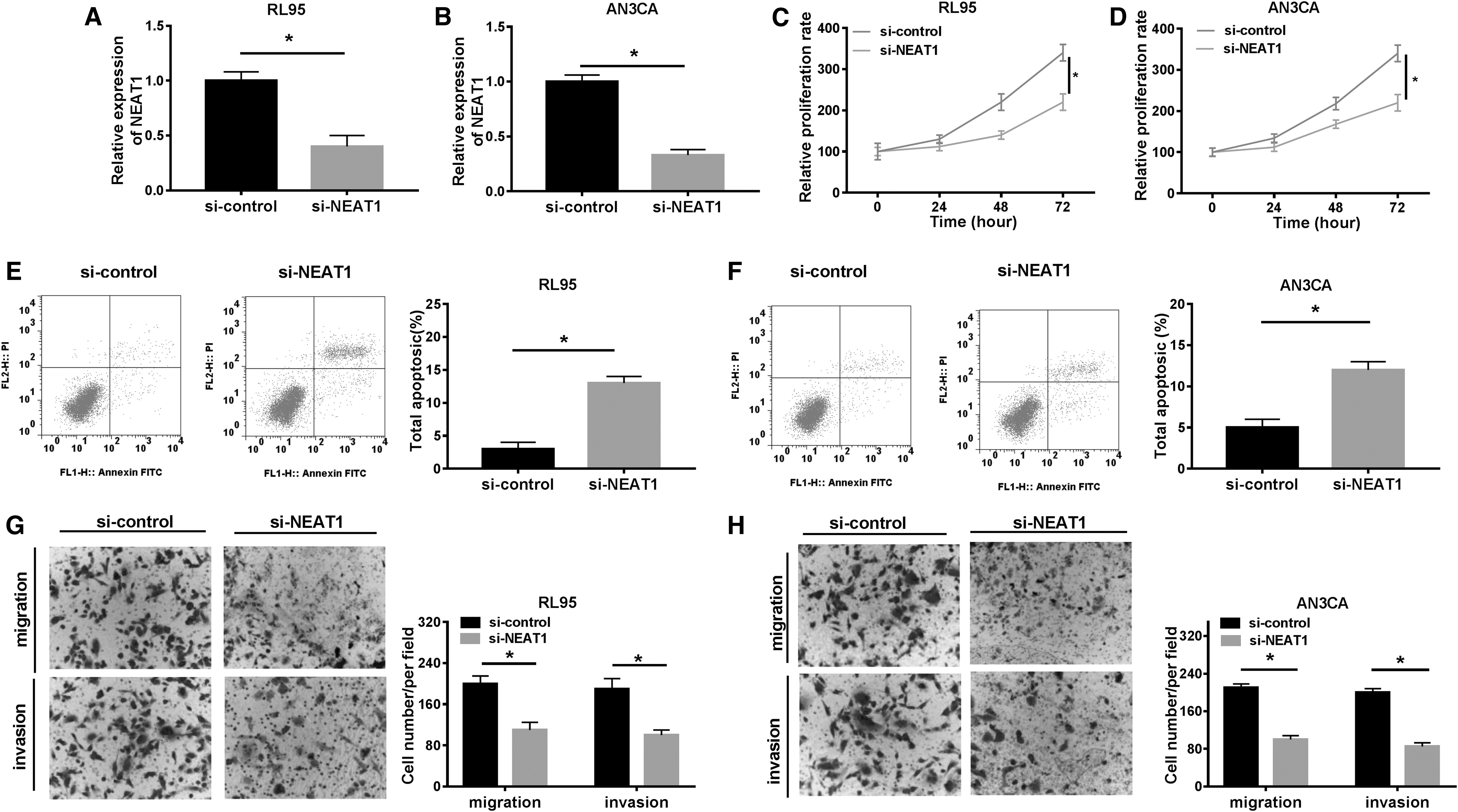
NEAT1 knockdown repressed cell proliferation, migration, and invasion in EC. RL95 and AN3CA cells were transfected with si-NEAT1 and si-control.
NEAT1 acted as a sponger of miR-202-3p
Bioinformatics analysis by online database starbase predicted that miR-202-3p had potential binding sites of NEAT1 (Fig. 3A). To validate the prediction, NEAT1-WT and NEAT1-MUT were co-transfected in RL95 and AN3CA cells with miR-202-3p or miR-NC. As displayed in Figure 3B and C, miR-202-3p reduced the luciferase activity of NEAT1-WT in EC cells. Moreover, miR-202-3p expression was enhanced by NEAT1 silencing in RL95 and AN3CA cells (Fig. 3D). More importantly, downregulated miR-202-3p was observed in EC tumors and cells compared with their counterparts (Fig. 3E, F). All the data proved that NEAT1 was a sponger of miR-202-3p in EC.

MiR-202-3p was directly interacted with NEAT1.
NEAT1 modulated EC cell progression by sponging miR-202-3p
RL95 and AN3CA cells were transfected with si-control, si-NEAT1, si-NEAT1+anti-miR-NC, and si-NEAT1+anti-miR-202-3p to explore the influences of NEAT1/miR-202-3p axis on EC cell development. Interestingly, miR-202-3p level in RL95 and AN3CA cells was increased after si-NEAT1 transfection and decreased after anti-miR-202-3p transfection (Fig. 4A, B). CCK-8 results demonstrated that NEAT1 silencing-induced suppressive effect on cell proliferation was restored by miR-202-3p inhibitor (Fig. 4C, D). Consistently, abundance of miR-202-3p restricted cell migration and invasion. However, elimination of miR-202-3p reversed the effects (Fig. 4E, F). Oppositely, NEAT1 silencing-induced apoptosis was blocked by miR-202-3p inhibitor (Fig. 4G, H). Altogether, miR-202-3p inhibitor abolished NEAT1 silencing-mediated effects on cell progression in EC.
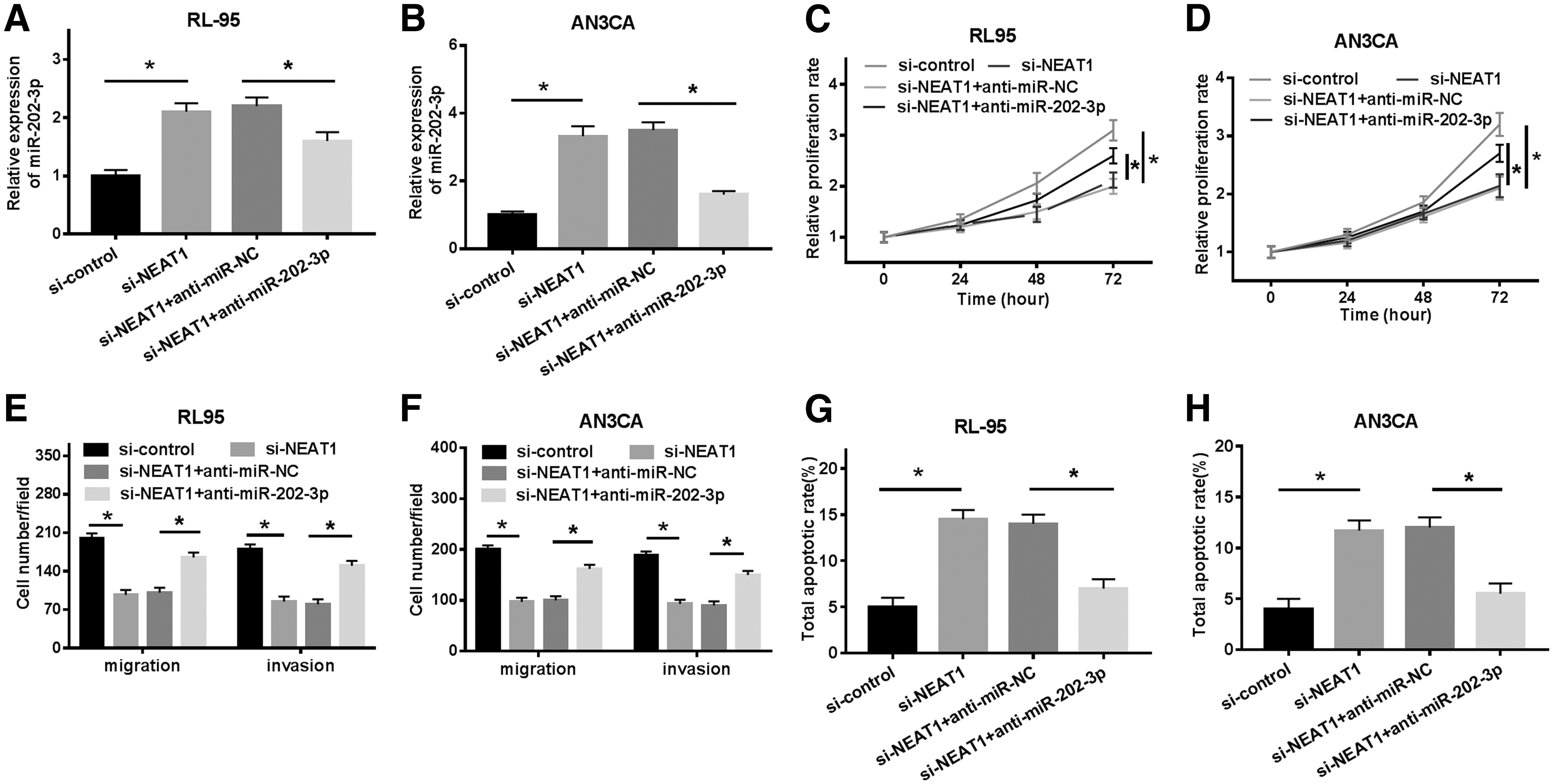
Inhibiting of MiR-202-3p attenuated NEAT1 knockdown-mediated effects on EC cells. RL95 and AN3CA cells were transfected with si-control, si-NEAT1, si-NEAT1+anti-miR-NC, and si-NEAT1+anti-miR-202-3p.
TIMD4 was targeted by miR-202-3p
By searching from Targetscan, miR-202-3p was found to comprise the binding sites of 3′-untranslated regions of TIMD4 (Fig. 5A). Reduction of luciferase activity in RL95 and AN3CA cells co-transfected with TIMD4-WT and miR-202-3p validated that miR-202-3p was directly interacted with TIMD4 (Fig. 5B, C). Furthermore, TIMD4 protein level was significantly decreased by miR-202-3p in RL95 and AN3CA cells (Fig. 5D, E). These findings clarified that TIMD4 was targeted by miR-202-3p in EC.
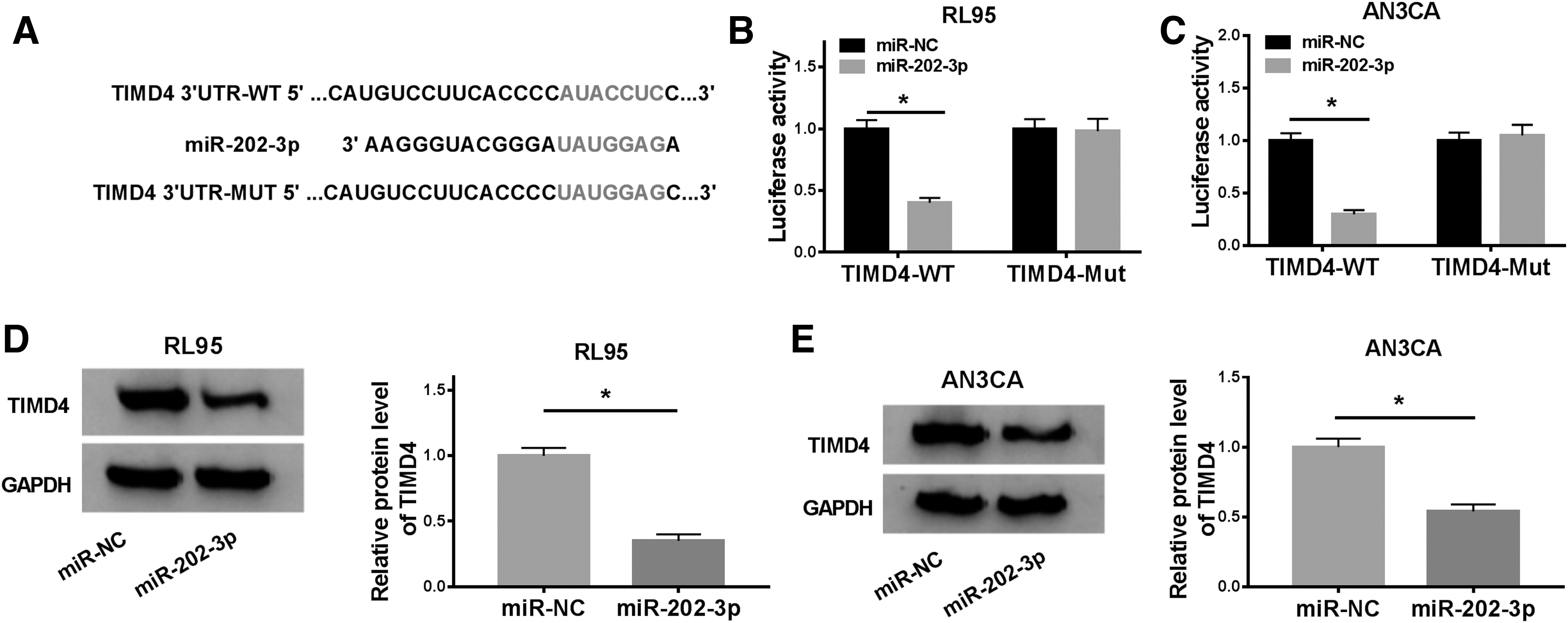
TIMD4 was a target of miR-202-3p.
Restoration of TIMD4 counteracted miR-202-3p mediated inhibition on cell progression in EC
To investigate the effects of miR-202-3p/TIMD4 axis on EC cell growth, miR-NC, miR-202-3p, miR-202-3p+pcDNA, and miR-202-3p+TIMD4 was transfected into RL95 and AN3CA cells. TIMD4 level in RL95 and AN3CA cells was reduced by miR-202-3p and enhanced by TIMD4 (Fig. 6A, B). As expected, upregulation of TIMD4 accelerated, whereas downregulation of TIMD4 attenuated EC cell proliferation (Fig. 6C, D). By contrast, cell apoptosis promoted by miR-202-3p was reversed by TIMD4 (Fig. 6E). Moreover, TIMD4 restored the suppressed cell migration and invasion caused by miR-202-3p (Fig. 6F, G). Likewise, the colonies exhibited the same trend (Fig. 6H). Hence, it is demonstrated that miR-202-3p regulated cell progression by targeting TIMD4 in EC.
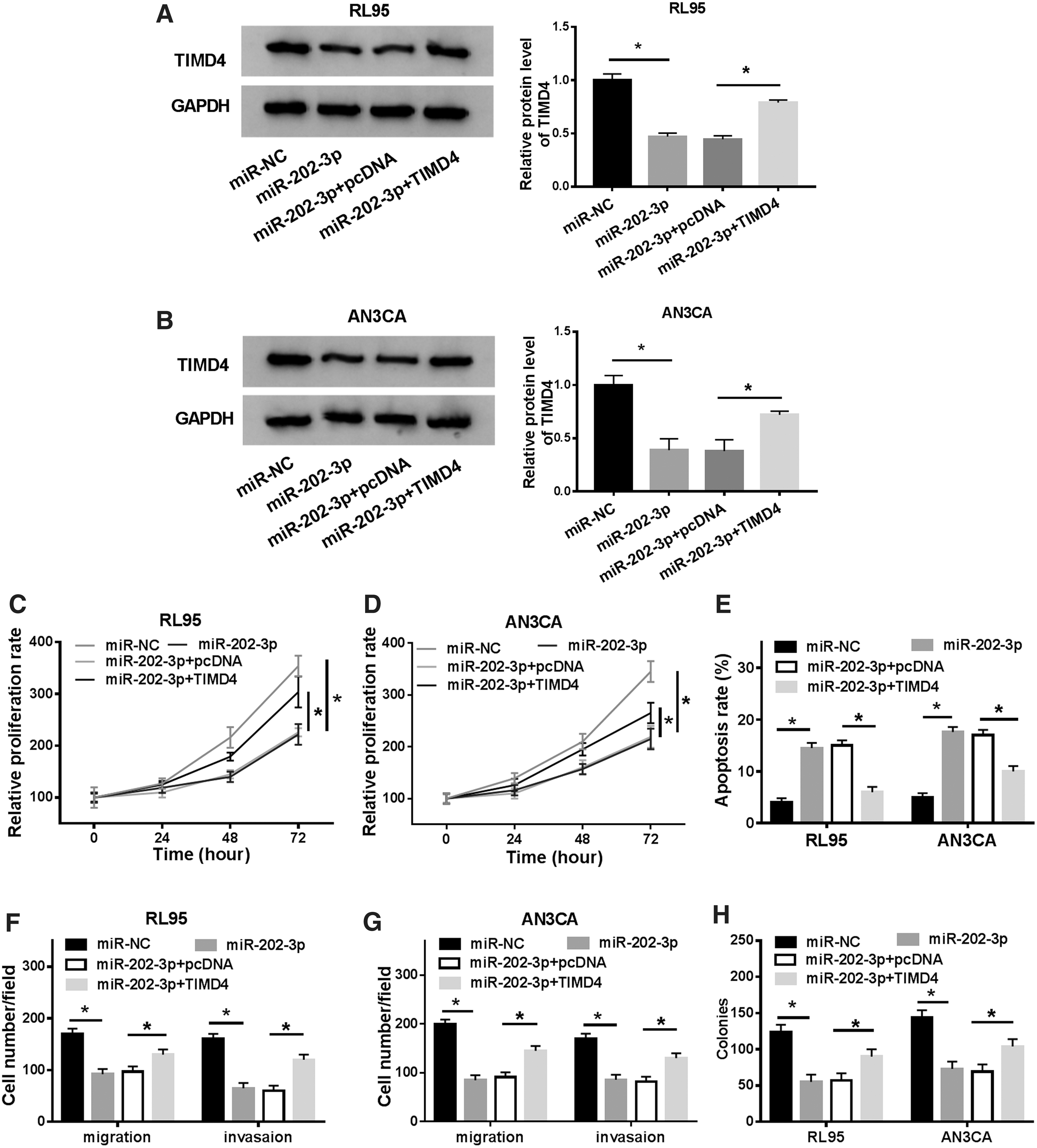
TIMD4 abrogated miR-202-3p mediated inhibitory effect on EC cells. RL95 and AN3CA cells were transfected with miR-NC, miR-202-3p, miR-202-3p+pcDNA and miR-202-3p+TIMD4.
NEAT1 affected regulated cell growth through miR-202-3p/TIMD4 axis in EC
The regulatory mechanism of NEAT1/miR-202-3p/TIMD4 axis was further studied. As illustrated in Figure 7A and B, TIMD4 mRNA expression was decreased by NEAT1 silencing, whereas increased by anti-miR-202-3p. Meanwhile, miR-202-3p silencing attenuated the inhibition induced by NEAT1 silencing on TIMD4 protein expression in EC (Fig. 7C, D). Collectively, NEAT1 was able to target miR-202-3p to regulate TIMD4 in EC.

NEAT1 regulated TIMD4 expression by sponging miR-202-3p. RL95 and AN3CA cells were transfected with si-control, si-NEAT1, si-NEAT1+anti-miR-NC, and si-NEAT1+anti-miR-202-3p.
Discussion
Recently, numerous evidences proved that dysregulation expression of lncRNA NEAT1 was a critical prognostic biomarker in many diseases, such as Alzheimer, liver injury, and nasopharyngeal carcinoma. 26 –28 For example, NEAT1 contributed to cell progression in pancreatic ductal adenocarcinoma by interacting with miR-302a-3p to modulate RELA proto-oncogene. 29 Increased expression of NEAT1 was reported to accelerate proliferation and repressed apoptosis in colon cancer through targeting miR-495-3p/CDK6 axis. 30 Consistently, Wang and colleagues revealed that NEAT1 contributed to hemangioma progression through binding to miR-361-5p to regulated vascular endothelial growth factor A. 31 In addition, NEAT1 interacted with miR-34a-5p to drive nasopharyngeal carcinoma cell survival, migration, invasion, and EMT by modulation of Wnt/β-catenin signaling. 32 Furthermore, NEAT1 activated EC cell viability, migration, and invasion. 16,32 –34 Consistent with the previous study, it is found that NEAT1 was upregulated in EC tissues and cells (RL95 and AN3CA). Furthermore, the knockdown of NEAT1 inhibited cell proliferation, migration, and invasion, and induced apoptosis in vitro.
Accumulated studies have demonstrated that lncRNAs are capable of regulating gene expression through targeting miRNAs. 35 –37 As predicted by starbase, it is indicated that miR-202-3p had potential binding sites of NEAT1. Also, the relationship between NEAT1 and miR-202-3p was verified by luciferase reporter assay. Aberrant expression of miR-202-3p was the pathogenesis in various diseases. 38 For instance, upregulated miR-202-3p could decrease myocardial ischemia-reperfusion injury through stimulating transforming growth factor beta 1/Smads signaling. 39 Interestingly, miR-202-3p was implicated in nonobstructive azoospermia by participating in sertoli cell progression and synthesis function regulation. 40 Moreover, miR-202-3p was reported to suppress tumor development by targeting ARL5A in human colorectal carcinoma. 41 In this study, miR-202-3p was downregulated in EC tissues and cell lines. Interestingly, miR-202-3p inhibitor could reverse the effects of NEAT1 knockdown on cell proliferation, migration, invasion, and apoptosis. Therefore, NEAT1 regulated EC cell growth through sponging miR-202-3p.
The abnormal protein expression of oncogene plays a crucial role in the progression of cancers. Thus, the authors further explored the downstream proteins regulated by miR-202-3p in EC. The online software Targetscan revealed that TIMD4 had the binding sites of miR-202-3p. And the interaction between them was verified by dual-luciferase reporter assay. TIMD4 is a type of TIMD family, which is mainly in antigen presenting cells. Previous reports showed that the TIMD4 was related to various malignant cancers. 42,43 The findings were in agreement with the previously described that TIMD4 acted as an oncogene in EC. In addition, there were inverse correlation between TIMD4 and miR-202-3p, and TIMD4 overexpression neutralized miR-202-3p-induced inhibitory effects on EC cells. In addition, NEAT1 enhanced TIMD4 expression through miR-202-3p in EC. Although the limitation of this study is the lack of in vivo experiments, a novel regulatory mechanism of NEAT1/miR-202-3p/TIMD4 might provide the new directions for further study in EC.
Conclusions
In summary, it is elucidated that NEAT1 facilitated cell proliferation, migration, invasion, and inhibited apoptosis through binding to miR-202-3p to upregulate TIMD4 expression in EC. These results might provide prospective markers for the diagnosis and treatment of EC.
Footnotes
Authors' Contributions
C.X. was in charge of project development, data collection, data analysis, and article writing. J.Z. was responsible for project development, data collection, data analysis, and article editing. Y.F. did data collection and data analysis.
Disclosure Statement
The authors have no conflict of interest to declare.
Funding Information
This research received no specific fund from any funding agency in the public and commercial.