
Abstract
Background:
This study intended to investigate the mechanisms underlying the epidermal growth factor receptor (EGFR) mutations in nonsmall cell lung cancer (NSCLC).
Materials and Methods:
Lung cancer tissue samples were collected from 20 patients with NSCLC (6 EGFR mutation types assigned into 2 categories and 14 EGFR wild types assigned to 4 categories). The samples were subjected to transcriptome sequencing, followed by identification of the differentially expressed mRNAs (DEMs), differentially expressed lncRNAs (DELs), and differentially expressed circRNAs (DECs) between the mutation and nonmutation groups. Function analysis and microRNA (miRNA) prediction for DEMs were performed. The correlations between long noncoding RNA (lncRNA)/circular RNA (circRNA) and messenger RNA (mRNA) were analyzed. In addition, the targeting lncRNA and circRNA of miRNA were predicted. Finally, competing endogenous RNA (ceRNA) network was constructed, and survival analysis for the mRNAs involved in the network was performed.
Results:
In total, 323 DEMs, 284 DELs, and 224 DECs were identified between EGFR mutation and nonmutation groups. The DEMs were significantly involved in gene ontology functions related to cilium morphogenesis and assembly. ceRNA networks were constructed based on the DEMs, DELs, DECs, and predicted miRNAs. Survival analysis showed that four genes in the ceRNA network, including ABCA3, ATL2, VAMP1, and APLN, were significantly associated with prognosis. The four genes were involved in several ceRNA pathways, including RP1-191J18/circ_000373/miR-520a-5p/ABCA3, RP5-1014D13/let-7i-5p/ATL2, circ_000373/miR-1293/VAMP1, and RP1-191J18/circ_000373/miR-378a-5p/APLN.
Conclusion:
EGFR mutations in NSCLC may be associated with cilium dysfunction and complex ceRNA regulatory mechanisms. The key RNAs in the ceRNA network may be used as promising biomarkers for predicting EGFR mutations in NSCLC.
Introduction
Lung cancer is a leading cause of cancer-related deaths worldwide. 1 Among the lung cancer patients, more than 70% have advanced nonsmall cell lung cancer (NSCLC). 2 Targeting the epidermal growth factor receptor (EGFR) is a promising strategy for treating NSCLC. Gefitinib and erlotinib are two widely used EGFR tyrosine kinase inhibitors, which are effective for treating advanced NSCLC. 3,4 However, the efficacy of the EGFR tyrosine kinase inhibitors is not consistent between patients. 5,6 Therefore, exploring valid predictive factors for improving the efficacy of EGFR tyrosine kinase inhibitors is significant for patients who may benefit more from this treatment.
The presence of somatic mutations in the kinase domain of EGFR was reported in 2004, and these mutations were demonstrated to be correlated with enhanced responsiveness to EGFR tyrosine kinase inhibitors in NSCLC patients. 7,8 Since then, EGFR-activating mutations have been confirmed to be good predictors of the efficacy of EGFR tyrosine kinase inhibitor treatment. 9,10 However, it was found that the patients with EGFR mutations were initially responsive to EGFR tyrosine kinase inhibitors, but eventually developed acquired resistance. 11,12 Unfortunately, the optimal treatment for patients with acquired resistance is still unclear. 11,12 Therefore, it is necessary to investigate the mechanisms underlying EGFR mutations.
Genome sequencing projects have revealed that over 90% of the human genome consists of noncoding RNAs. 13 MicroRNAs (miRNAs) are endogenous small noncoding RNAs, which have been identified as key players in various pathological conditions, including human cancers. 14 –16 Long noncoding RNAs (lncRNAs) represent a class of noncoding RNAs longer than 200 nucleotides, 17 which are also implicated in cancer progression. 18 Circular RNAs (circRNAs) are novel endogenous noncoding RNAs, which play an important role in the initiation and progression of human diseases, especially cancers. 19 A recent study identified a new regulatory circuitry, where RNAs crosstalk with each other by competing for shared miRNAs. These competing endogenous RNAs (ceRNAs) can regulate the distribution of miRNAs on their targets, thereby imposing an additional level of post-transcriptional regulation. 20 –22 A previous study reported that both lncRNAs and circRNAs could serve as ceRNAs to sequester miRNAs. 23 Importantly, ceRNAs are involved in the pathogenesis of several common cancers, including lung cancer. 23 Nevertheless, to the authors' best knowledge, the potential role of ceRNAs has not been investigated in EGFR mutations in NSCLC.
Thus, this study aimed to investigate the mechanisms underlying EGFR mutations in NSCLC at the transcriptome level and explore the potential regulatory mechanisms in the context of ceRNAs.
Materials and Methods
Tissue sample collection
Lung cancer tissues were collected from 8 patients with squamous cell lung cancer (2 EGFR mutation types and 6 EGFR wild types) and 12 patients with lung adenocarcinoma (4 EGFR mutation types and 8 EGFR wild types) undergoing surgical removal of tumor at the Shaanxi Provincial People's Hospital between October 2015 and March 2017. Therefore, the experimental set consisted of six samples with EGFR mutations and 14 samples with wild-type EGFR. The histopathologic diagnosis of all the specimens was performed in accordance with the World Health Organization classification criteria and the Union for International Cancer Control-American Joint Committee on Cancer staging system based on tumor, node, metastasis classification for lung cancer. The cancer tissues were cut into tissue blocks (0.8 × 0.5 cm), rinsed repeatedly with sterile saline, and then stored at −80°C. Informed consent was obtained from all the patients and the samples were collected under an Institutional Review Board approved protocol in Shaanxi Provincial People's Hospital.
RNA isolation, library preparation and sequencing
The samples with EGFR mutations were assigned into two categories, and the EGFR wild-type samples were assigned into four categories. Total RNA was isolated from the six sample categories using TRIzol reagent (Invitrogen, Carlsbad, CA), and the integrity of the RNA was evaluated on an Agilent Bioanalyzer 2100 (Agilent Technologies, USA). The ribosomal RNAs (rRNAs) were removed using the Ribo-zero™ rRNA Removal Kit (Epicenter, USA). The RNA was then broken into fragments (200–300 bp) using divalent cations, and the first-strand complementary DNA (cDNA) was synthesized using M-MuLV reverse transcriptase and random primers (Invitrogen). Subsequently, the second-strand cDNA was synthesized using DNA polymerase I and RNase H. The dTTP in the reaction buffer were replaced with dUTP. cDNA fragments of 300–400 bp were selected for PCR to obtain an enriched cDNA library. The PCR products were assessed using an Agilent Bioanalyzer 2100 system. Finally, the libraries were sequenced on an Illumina HiSeq 2500 platform.
Raw data processing and quality control
For quality control of raw data, the adapter sequences in the raw reads were removed using Cutadapt (version 1.2.1) with at least 10 bp overlap, and the mean quality value of each window was controlled to be at least 20 to obtain high-quality clean data.
Differentially expressed mRNA/lncRNA/circRNA analysis
Differences between the EGFR mutation and wild-type (nonmutation) groups were analyzed. The p-value was calculated using the nonpaired t-test in limma package (version 3.30.13) 24,25 of R (version 3.3.2), and then adjusted with Benjamini–Hochberg method. The differentially expressed mRNAs (DEMs), differentially expressed lncRNAs (DELs), and differentially expressed circRNAs (DECs) between the two groups with thresholds of |log fold change| > 0.585 and p-value <0.05 were considered statistically significant.
Protein–protein interaction network analysis
The interactions between the DEMs encoding proteins were predicted and analyzed using the Search Tool for the Retrieval of Interacting Gene 26 database. The parameter setting was protein–protein interaction (PPI) score = 0.4 (median confidence). Based on the obtained interaction pairs, the PPI network was constructed using Cytoscape (version 3.6.0) 27 software. The key nodes involved in the PPI network were identified.
Functional enrichment analysis
To identify the possible enrichment results of the DEMs, Gene Ontology (GO) 28 functional enrichment analyses were carried out using Database for Annotation Visualization and Integrated Discovery. 29 The GO terms consisted of three categories: biological process, molecular function, and cellular component. Results with count ≥2 and p < 0.05 were considered statistically significant enrichments.
Co-expression analysis
The correlations between lncRNA/circRNA and messenger RNA (mRNA) were analyzed using Pearson correlation coefficient method, and the co-expression relation pairs of lncRNA-mRNA and circRNA-mRNA were predicted with p < 0.01 and correlation coefficient |r| > 0.95. Among these co-expression relation pairs, the pairs with r > 0.95 were positively correlated pairs and with r < −0.95 were negatively correlated pairs.
miRNA prediction
The targeting miRNAs of the DEMs were predicted using miRWalk2.0 database. 30,31 The databases and tools in miRWalk2.0 included miRWalk, miRMap, miRanda, miRNAMap, RNA22, Pictar2, PITA, and Targetscan. Finally, the miRNAs predicted by seven of the eight databases or tools were retained.
In general, lncRNAs and circRNAs compete to bind to miRNAs, thereby relieving the inhibitory effect of the miRNAs on their target genes, leading to increased expression of the target genes. Therefore, the authors focused on the positive correlations of co-expression of lncRNA-mRNA and circRNA-mRNA. The lncRNAs and circRNAs that were involved in positively correlated lncRNA-mRNA and circRNA-mRNA pairs were selected for miRNA targeting lncRNA and circRNA prediction. Briefly, based on the miRNA, lncRNA, and circRNA sequence files, the targeting lncRNA and circRNA of miRNA were predicted using miRanda (version 3.3a). 32 The parameters were set as -sc 160, -en-20, and -strict.
Pathway enrichment analysis for miRNA/lncRNA/circRNA
The miRNAs and Kyoto Encyclopedia of Genes and Genomes (KEGG) [18] pathways were analyzed based on their target genes using clusterProfile package (version 3.2.14). 33 In addition, the KEGG pathways enriched by the lncRNAs and circRNAs were predicted based on the mRNAs involved in lncRNA/mRNA and circRNA/mRNA relation pairs using clusterProfile package as well. The pathways with p-value <0.05 were considered significant.
Construction of lncRNA/miRNA/mRNA and circRNA/miRNA/mRNA networks
Based on the predicted miRNA/target and miRNA/lncRNA pairs, as well as co-expression relation pairs of mRNA/lncRNA, the miRNAs that targeted both mRNA and lncRNA were screened out, and the lncRNA/miRNA/mRNA regulatory network was constructed using Cytoscape. Similarly, the miRNAs that targeted both mRNA and circRNA were selected as well to construct the circRNA/miRNA/mRNA regulatory network using Cytoscape.
ceRNA network analysis
Based on the lncRNA/miRNA/mRNA and circRNA/miRNA/mRNA regulatory networks, the miRNAs that targeted mRNAs, lncRNAs, and circRNAs at the same time were selected for the construction of the ceRNA network.
Survival prognosis analysis
The squamous cell lung cancer and lung adenocarcinoma data (clinical survival information and gene expression data) in The Cancer Genome Atlas (TCGA) database were downloaded for survival prognosis analysis of all mRNAs involved in the ceRNA network. Briefly, using the survival package 34 in R (version 2.41-3), the effect of DEMs (involved in the ceRNA regulatory network) on patient prognosis was analyzed, and the Kaplan–Meier survival curve was plotted followed by log-rank test. The genes with p < 0.05 were considered survival prognosis-related genes.
Results
Differential expression analysis
Following the analysis, a total of 323 DEMs, including 278 downregulated and 45 upregulated ones, were identified between the EGFR mutation and nonmutation groups. In addition, 284 DELs (170 downregulated and 114 upregulated) and 224 DECs (175 downregulated and 49 upregulated) were identified. Clustering heatmaps showed that these DEMs, DELs, and DECs were able to separate the EGFR mutation and nonmutation groups significantly (Fig. 1).
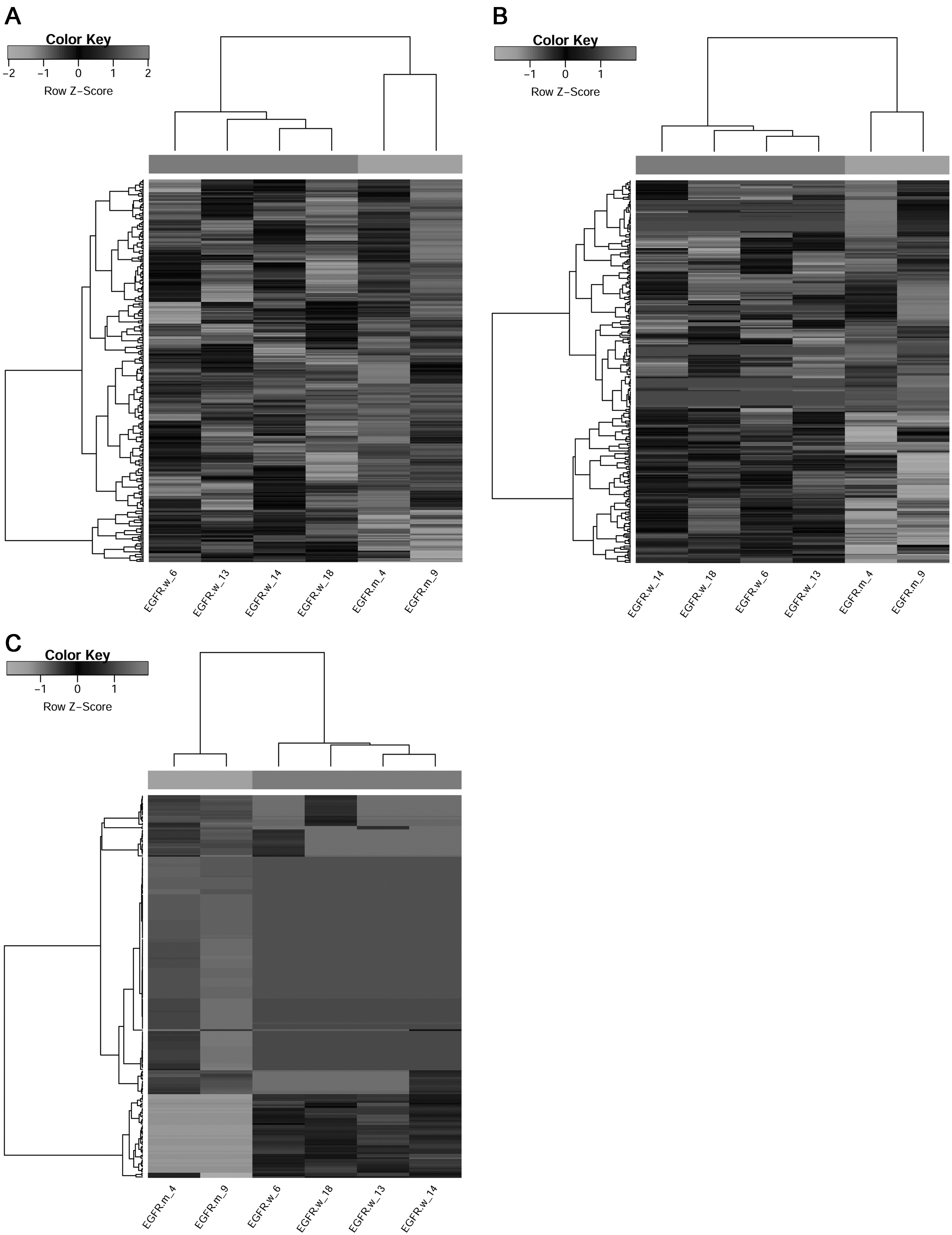
The clustering heatmaps of the differentially expressed mRNA
PPI network analysis
The PPI network consisted of 153 nodes and 172 interaction relation pairs. The nodes with higher topology scores (degree ≥5) were considered hub nodes, including the ATM serine/threonine kinase (ATM), regulator of nonsense-mediated mRNA decay (UPF3B), embryonic ectoderm development, and G elongation factor mitochondrial 2.
Functional enrichment analysis for DEMs
GO analysis showed that the DEMs were significantly enriched in 11 biological process terms, six cellular component terms and six molecular function terms, including cilium morphogenesis (KIF3A, CEP162, and TTBK2), cilium assembly, forebrain development (KIF3A, CEP162, and TTBK2), and apoptotic signaling pathway (TNFRSF11A, UACA, CASP8, and TICAM1) (Fig. 2).
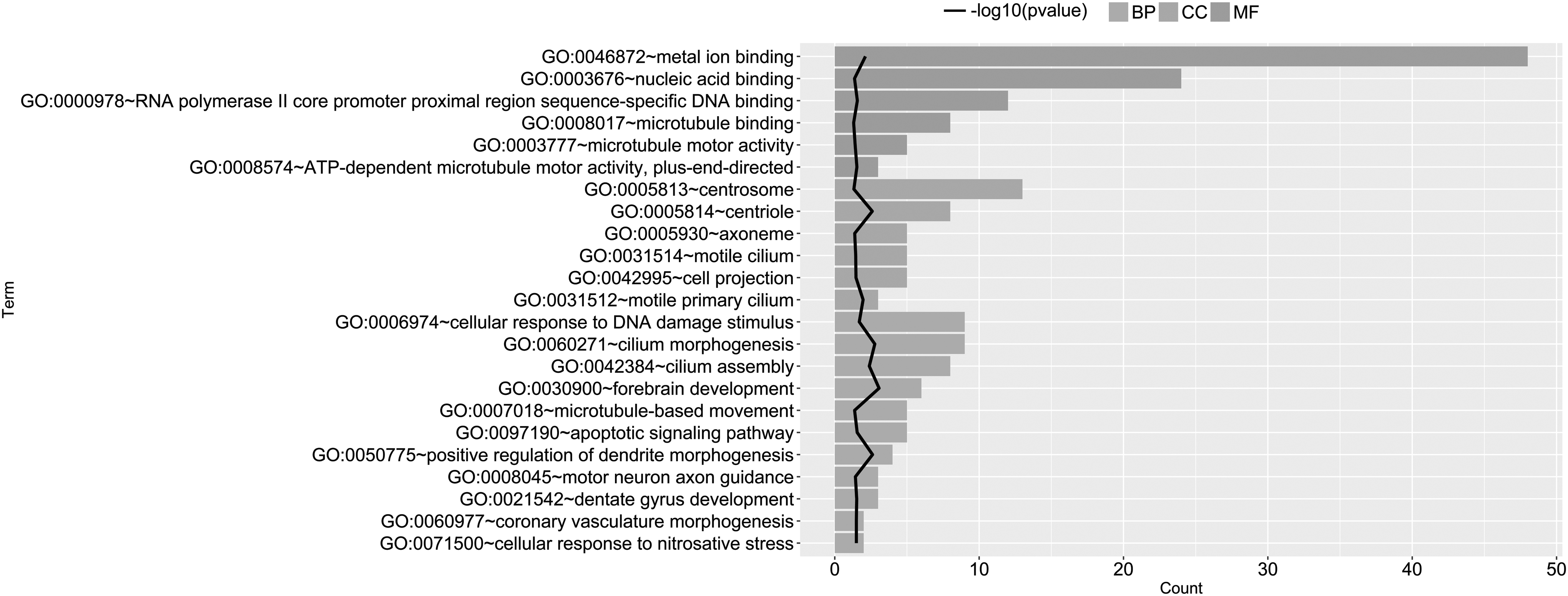
GO enrichment analysis of significantly differentially expressed mRNAs. The abscissa represents the number of genes enriched in each GO term and the ordinate represents the name of the GO term. The black broken line represents −log10 (p-value). BP, biological process; CC, cellular component; GO, Gene Ontology; MF, molecular function.
Co-expression analysis
Based on p < 0.01 and |r| > 0.95, a total of 4472 co-expressed lncRNA-mRNA pairs, involving 323 mRNAs and 54 lncRNAs, were predicted. Among these lncRNA-mRNA pairs, there were 2850 positively correlated pairs and 1622 negatively correlated pairs. Furthermore, 4942 co-expressed circRNA-mRNA pairs (3624 pairs with positive correlation and 1318 pairs with negative correlation) were predicted, involving 182 mRNAs and 224 circRNAs.
miRNA prediction and pathway analysis
A total of 200 miRNAs were predicted based on the DEMs. The miRNAs that targeted lncRNAs and circRNAs were also predicted and included 253 lncRNAs and 212 circRNAs. The identified miRNAs, lncRNAs, and circRNAs were subjected to KEGG pathway analysis. The miRNAs were significantly enriched in soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) interactions in vesicular transport (miR-1293), Apelin signaling pathway (miR-1237-3p, miR-124-3p, and miR-378a-5p), and fatty acid- and amino acid-associated pathways (miR-128-3p). The lncRNA, AC034186, was involved in the relaxin and Ras signaling pathways. The retinol metabolism pathway was enriched by several lncRNAs, including AC005740, AC007228, and AC020626. The circRNAs were involved in Ras signaling pathway (circ_004103), PI3K-Akt signaling pathway (circ_004103), and human papillomavirus infection (circ_012076 and circ_013487).
lncRNA/miRNA/mRNA network, circRNA/miRNA/mRNA network, and ceRNA network construction
The lncRNA/miRNA/mRNA network was constructed with 259 nodes and 518 relation pairs. In the circRNA/miRNA/mRNA network, there were 217 nodes and 390 relation pairs. The ceRNA network was constructed based on the two integrated networks, including 289 nodes and 631 relation pairs (Fig. 3). The mRNAs, miRNAs, lncRNAs, and circRNAs with top 5 degree values in the ceRNA network are shown in Table 1.

The ceRNA regulatory network. Red represents the upregulated nodes, green represents the downregulated nodes, and yellow represents the predicted miRNA, whose upregulation or downregulation is unclear. Circular node represents mRNA, hexagonal node represents miRNA, triangular node represents lncRNA, and arrow node represents circRNA. Node size represents the degree. ceRNA, competing endogenous RNA; miRNA, microRNA.
The Top 5 Messenger RNAs, Micro RNAs, Long Noncoding RNAs, and Circular RNAs in Competing Endogenous RNA Network Were, Respectively, Displayed According to Degree Values
circRNA, circular RNA; lncRNA, long noncoding RNA; miRNA, microRNA; mRNA, messenger RNA.
Survival prognosis analysis based on TCGA database
The expression data for all mRNAs (in the ceRNA network) in squamous cell lung cancer and lung adenocarcinoma samples were extracted from TCGA database. Survival prognosis analysis of all mRNAs involved in the ceRNA network was conducted based on the clinical information and the median value of mRNAs. Finally, only four mRNAs were found to be correlated to prognosis of patients. In detail, ATP-binding cassette subfamily A member 3 (ABCA3), atlastin GTPase 2 (ATL2), and vesicle-associated membrane protein 1 (VAMP1) were significantly associated with the prognosis of patients with lung adenocarcinoma; and Apelin (APLN) was significantly related with the prognosis of patients with squamous cell lung cancer. The Kaplan–Meier survival curves of the four genes are shown in Figure 4.

Kaplan–Meier survival curves of ABCA3
Discussion
In this study, a total of 323 DEMs, 284 DELs, and 224 DECs were identified between EGFR mutation and nonmutation groups. The DEMs were significantly involved in GO functions related to cilium morphogenesis and assembly. Survival analysis revealed that four genes in the ceRNA network, including ABCA3, ATL2, VAMP1, and APLN, were significantly related with the prognosis of NSCLC patients. The four genes were involved in several ceRNA pathways, such as RP1-191J18/circ_000373/miR-520a-5p/ABCA3, RP5-1014D13/let-7i-5p/ATL2, circ_000373/miR-1293/VAMP1, and RP1-191J18/circ_000373/miR-378a-5p/APLN.
The airway epithelium contains four major cell types: ciliated, secretory, undifferentiated intermediate, and basal cells. 35 The ciliated cells are the first line of defense against inhaled particulates and pathogens. Dysfunction of the mucociliary escalator can weaken the defenses of the epithelium, leading to lung disease. 36 A previous study reported that the primary cilium played an essential role in regulating cellular responses to Shh and PDGF signaling, two important pathways in cancer biology, 37 which indicated the important role of primary cilium in carcinogenesis. In this study, the DEMs such as kinesin family member 3a (KIF3A), centrosomal protein 162 (CEP162), and tau tubulin kinase 2 (TTBK2) were significantly involved in GO functions related to cilium morphogenesis and cilium assembly, suggesting that EGFR mutations in NSCLC might be associated with cilium dysfunction.
ceRNAs are RNAs that share sequences called miRNA recognition elements, which are recognized by the miRNAs. The ceRNAs act as important regulators of gene expression by sponging miRNAs. 20 –22 In this study, the authors constructed ceRNA networks based on DEMs, DELs, DECs, and predicted miRNAs to explore the potential regulatory mechanism of EGFR mutations in NSCLC. Furthermore, they performed survival analysis for the mRNAs involved in the ceRNA network and identified four mRNAs (ABCA3, ATL2, VAMP1, and APLN) that were significantly correlated with the prognosis of NSCLC patients. Therefore, the authors focused on the four genes involved the ceRNA pathways, such as RP1-191J18/circ_000373/miR-520a-5p/ABCA3, RP5-1014D13/let-7i-5p/ATL2, circ_000373/miR-1293/VAMP1, and RP1-191J18/circ_000373/miR-378a-5p/APLN.
ABCA3 is a member of the superfamily of ATP-binding cassette transporters, which is recognized as a regulatory transport protein during the process of surfactant extrusion from type II pneumocytes into the pulmonary alveolae. 38,39 A recent study reported that ABCA3 was a prognostic factor for lung cancer. 40 In accordance with these findings, the results showed that ABCA3 was significantly associated with the prognosis of patients with lung adenocarcinoma. Importantly, Overbeck et al. 41 demonstrated that the ATP-binding cassette transporter proteins, including ABCA3, are involved in drug resistance in lung cancer cell lines. As the authors mentioned above, patients with EGFR mutations eventually develop acquired resistance. 11,12 Therefore, they speculate that RP1-191J18 and circ_000373 may serve as ceRNAs to regulate ABCA3 expression and affect drug resistance in NSCLC with EGFR mutation.
In the ceRNA pathway of circ_000373/miR-1293/VAMP1, miR-1293 was involved in the pathway of SNARE interactions in vesicular transport. SNAREs are critical proteins that mediate many cellular processes, such as cell growth, synaptic vesicle fusion, and protein transport. Studies have reported that SNAREs play an important role in cancer progression. Importantly, SNAREs can transport molecules conferring chemotherapy resistance. 42 In ceRNA pathway of RP5-1014D13/let-7i-5p/ATL2, let-7i was found to be upregulated in lung adenocarcinoma. 43 Let-7i-5p has been identified as a tumor suppressor, the upregulation of which markedly inhibited colon tumor cell viability and invasion in colon cancer. 44 However, to the authors' knowledge, the functions of the two ceRNAs in lung cancer with EGFR mutation have not been reported. The authors speculate that EGFR may become mutated in NSCLC through RP5-1014D13/let-7i-5p/ATL2 and circ_000373/miR-1293/VAMP1.
Apelin (APLN) is an endogenous ligand of the apelin receptor. 45 APLN has been reported to be upregulated in NSCLC tissues compared with normal lung tissues, and high expression level of APLN protein is associated with poor overall survival. 46 In addition, overexpression of APLN in NSCLC cells stimulated tumor growth, tumor cell proliferation, and microvessel densities and perimeters. Interestingly, in the ceRNA pathway of RP1-191J18/circ_000373/miR-378a-5p/APLN, miR-378a-5p was involved in the apelin signaling pathway, further suggesting the important role of apelin in NSCLC with EGFR mutation.
However, the authors did not perform an experimental validation to confirm these findings. In further studies, it is valuable to obtain clinical cases, conduct cohort studies, and observe overall survival and clinical outcome after treatment.
In conclusion, the authors' findings reveal that EGFR mutations in NSCLC may be associated with cilium dysfunction, as well as complex ceRNA regulatory mechanisms. These key RNAs in the ceRNA regulatory network may be used as promising biomarkers for predicting EGFR mutations in NSCLC.
Footnotes
Ethics Approval and Consent to Participate
This study was approved by Ethics Committee of Shaanxi Provincial People's Hospital and University of Arkansas for Medical Science.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
Authors' Contributions
Y.T. and L.S. conceived and designed the research. M.Y. and X.N. acquired the data. Y.T. and Q.N. analyzed and interpreted the data. M.Y., J.Y., and Y.L. performed statistical analysis. Y.T., S.H., and J.G. participated in the obtaining funding. Y.T. drafted the article. S.S. and F.L. revised the article for important intellectual content. All authors read and approved the final article.
Disclosure Statement
The authors declare that there is no conflict of interests.
Funding Information
This work was supported by Shaanxi Province Natural Science Research Project (Program Nos. 2018JM7044, 2019SF-185); Key Projects of Social Development Research Programs of Shaanxi Province (Program No. 2017SF-199); Research foundation of Shaanxi Provincial Administration of traditional Chinese Medicine (Program No. JCPT039); Xi'an Science and Technology Bureau of social development projects [Program Nos. 2016047SF/YX03(4); 201805093YX1SF27(9)]; Xi'an Medical College Advanced Subjects Project Funded (Program No. 16SXK); and Shaanxi Province Health Family Planning Commission Research Projects (Program Nos. 2016D036; 2018D019).