
Abstract
Objective:
This study aimed to identify the novel microRNAs (miRNAs) for early diagnosis of bladder cancer.
Materials and Methods:
Differentially expressed miRNAs between early and advanced bladder cancer were identified by differential expression analysis, using miRNA-seq data from The Cancer Genome Atlas (TCGA). The optimal subset of feature miRNAs for pathologic stage prediction was acquired by Random Forest algorithm and was used to construct a support vector machine (SVM) classifier. The performance of the SVM classifier in predicting the progression of bladder cancer samples was validated using an independent validating dataset. An miRNA-regulated target gene network was finally constructed and functional annotation were performed for the target genes.
Results:
A total of 52 significantly differentially expressed miRNAs were identified between early and advanced bladder cancer samples and 17 of these miRNAs were identified to be feature miRNAs. The 17 feature miRNAs were used to construct an SVM classifier, which showed a high performance in pathologic stage prediction for both training and validating dataset. Besides, our functional annotation analysis showed that the feature miRNAs were significantly involved in biological processes and pathways related to extracellular matrix process and PI3K/Akt signaling.
Conclusions:
The optimal subset of miRNAs may act as potential therapeutic targets and diagnostic markers of bladder cancer.
Introduction
As one of several urological cancers, bladder cancer (BC) is the sixth most common cancer, with an estimated 79,030 new cases and 16,870 deaths in the United States in 2017. 1 Although bladder cancer from the majority (75%–85%) of primary patients was diagnosed as nonmuscle invasive bladder cancer, the recurrence rate after transurethral resection can be up to 75% within 5 years. 2,3 When the bladder cancer becomes more invasive to the muscle layer, it results in poor clinical outcome with a 5-year survival rate of ∼57%. 4 Therefore, investigation on bladder cancer, especially on the mechanisms of its progression, will help to advance clinical diagnosis and treatment of this disease.
MicroRNAs (miRNAs) are a large family of highly conserved noncoding RNAs with ∼22 nucleotides in length, which post-transcriptionally regulate gene expression by directly targeting mRNAs. 5,6 MiRNAs have been shown to control cell growth, differentiation, and apoptosis. 7 Moreover, miRNAs are significantly deregulated in many cancers types. 8,9 They may serve as either oncogenes or tumor suppressors, and play important roles in the oncogenesis and progression of various carcinomas, including bladder cancer. 10,11 Increasing evidence has shown that miRNA expression profile is significantly altered in tumor specimens of bladder cancer patients. 12,13 However, investigation on identification of miRNA expression profile during bladder cancer progression is limited.
Machine learning has undergone significant development over the past years and is being used successfully in many intelligent applications covering a wide array of data-related problems. Recently, machine learning is being applied for diagnosis in medical regions. 14,15 As the cost of next-generation sequencing (NGS) is becoming lower, thousands of samples are investigated by NGS. Researchers have identified many miRNAs as biomarkers for many types of cancer, such as miRNA-200c, 16 miRNA-let7, 17 and so on. However, more comprehensive analysis of miRNA screening is limited. Machine learning makes it possible.
At present, the Cancer Genome Atlas (TCGA) database provides comprehensive cancer datasets including DNA-seq, mRNA-seq, and miRNA-seq for researchers. In this study, we reanalyzed the data about bladder cancer from the TCGA database and identified the feature miRNAs that can predict the development of bladder cancer. Besides, based on the feature miRNA, we constructed a support vector machine (SVM) classifier, which showed a high performance in pathologic stage prediction of bladder cancer. These findings may act as important therapeutic prognostic markers in progression of bladder cancer and improve bladder cancer treatment.
Materials and Methods
Data source and preprocessing
The mRNA and miRNA expressing data (Illumina HiSeq2000) of bladder cancer were downloaded from the TCGA database (
Group Information
The Detail Information of the Training Set and Validating Set
CR, complete response; NA indicates the missing value; p was calculated by chi-square test; PD, progressive disease; PR, partial response; SD, stable disease.
Screening of differentially expressed miRNAs between different pathologic phases
The raw count of all miRNA-seq data were normalized using the trimmed mean of M-values method (TMM) algorithm from edgeR package in R (Version: 3.4) and the mean–variance relationship was modeled using the precision weights (voom) method from edgeR package. Then differential expression analysis of miRNA data was performed using linear regression and empirical Bayesian methods from limma package. 20 p-Values were adjusted for multiple testing using the Benjamini and Hochberg false discovery rate, and differently expressed miRNAs were considered when adjusted p < 0.05 and fold change was >1.5. Besides, the differential expression analysis was also performed between NMIBC versus NMIBC_normal, MIBC versus MIBC_normal utilizing the limma package. 21
miRNA cluster analysis
Bidirectional hierarchical clustering based on the centered Pearson correlation algorithm 22 was performed on differentially expressed miRNAs. The correlation between the resulting clusters and the distribution of samples with different pathologic stages were tested using chisq.test function of R. The correlation between the clusters and prognosis were calculated using the Kaplan–Meier (KM) algorithm from survival package in R. 23
Screening of feature miRNAs
Feature miRNAs were selected from differentially expressed miRNAs using the bootstrap algorithm of the Random Forest package under R. The optimal subset of feature miRNAs was obtained when out-of-bag (OOB) error rate reached the minimum.
Construction of SVM classifier
An SVM classifier was constructed based on the feature miRNAs using the SVM method (core function: Sigmoid Kernel; cross: 10 times cross-validation) of the R package e1071. 24 The samples were classified as early-stage-like and advanced-stage-like groups by the SVM classifier. The correlation between the SVM classification groups and prognosis were evaluated using the KM algorithm.
Validation analysis
The optimal subset of feature miRNAs was validated using validating dataset by two-way hierarchical clustering and SVM classification and KM survival analysis.
Functional analysis of miRNA-related genes
Differentially expressed mRNAs between different disease stages were screened by differential expression analysis method for miRNA. The differently expressed mRNAs targeted by the feature miRNAs were searched by miRanda (parameter: default score 140) and used to construct a feature miRNA-mRNA regulation network. Finally, all the miRNA-regulated mRNAs were subjected to Gene Ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses by DAVID 25 with the criterion of p < 0.05.
Results
Identification of significantly altered miRNAs in the progression of bladder cancer
In total, 52 significantly differentially expressed miRNAs between advanced- and early-stage groups were obtained (Fig. 1A). Bidirectional hierarchical clustering analysis of the differentially expressed miRNAs showed that the training samples were clearly divided into two categories: cluster 1 and cluster 2 (Fig. 1B). Cluster 1 was dominated by early-stage samples, including 62 early-stage samples and 56 advanced-stage samples. Cluster 2 was dominated by advanced-stage samples, including 119 advanced-stage samples and 29 early-stage samples. Specifically, 68.13% (62/91) of the early group was in cluster 1, and 68% (119/175) of the advanced group was in cluster 2. The overall accuracy rate of the classification was 68.04% (189/266). The chi-square test showed that the distribution of early- and advanced-stage samples were significantly different between the two clusters (χ 2 = 30.22, p = 3.858e-08), indicating that clusters were significantly correlated with the development of disease. Further KM survival analysis showed that cluster 1 had a better prognosis than cluster 2 (logRank p = 0.00052; Fig. 1C).
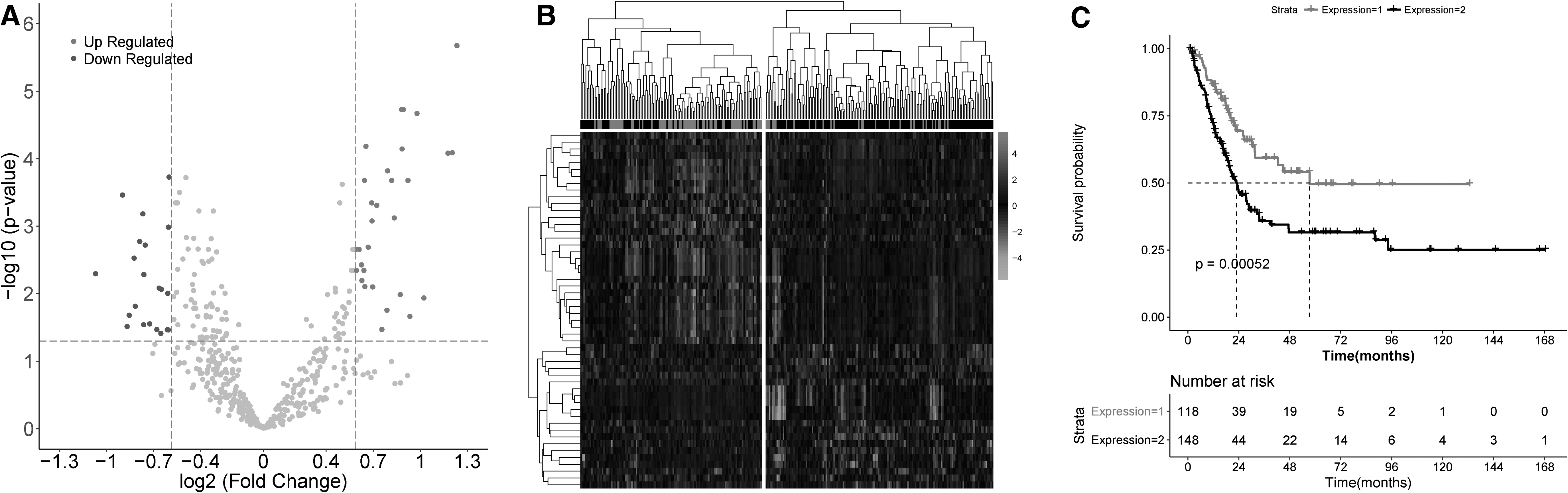
Identification of significantly altered miRNAs in the progression of bladder cancer.
Identification of feature miRNAs
The Random Forest algorithm was used to obtain the optimal subset of feature miRNAs from the 52 differentially expressed miRNAs. Our results showed that 17 miRNAs (Table 3) were used for fitting when the OOB error reached the minimum value 0.24 and considered as the optimal feature miRNA subset (Fig. 2A). Bidirectional hierarchical clustering based on the expression of 17 feature miRNAs showed that the samples were also clearly divided into two categories, cluster 1 and cluster 2 (Fig. 2B). Specifically, cluster 1 mainly consisted of early-stage samples, including 55 early- and 45 advanced-stage samples. Cluster 2 mainly consisted of advanced-stage samples, including 130 advanced- and 36 early-stage samples. The overall accuracy rate of the classification was 69.5% (185/266), similar to the results obtained using 52 differently expressed miRNAs. Moreover, the KM survival analysis showed that cluster 1 had a better survival prognosis than cluster 2 (logRank p = 0.0014, Fig. 2C).

Identification of feature miRNAs
17 Feature MicroRNAs Related to the Progression of Bladder Cancer
FC, fold change.
Construction of SVM classifier
Based on the optimal subset of feature miRNAs, an SVM classifier was constructed to predict different disease stage samples. Samples in the training dataset were divided into early-stage-like and advanced-stage-like groups. The results showed that the SVM classifier could accurately predict 226 of the 266 training samples. The overall accuracy rate was 85% (sensitivity = 98%, specificity = 60%, positive predictive value [PPV] = 83%, negative predictive value [NPV] = 93%) and the area under receiver operating characteristic (AUROC) was 0.914 (Fig. 3A). Besides, the KM survival analysis showed that the groups from SVM classifier were significantly correlated with prognosis (logRank p = 0.0011; Fig. 3B).

Construction of SVM classifier
Validation of the optimal subset of feature miRNAs
The performance of the 17 feature miRNAs was validated using validating dataset. Bidirectional hierarchical clustering based on the expression value of the 17 miRNAs showed that the samples in the validating dataset could be divided into two groups, cluster 1 and 2 (Fig. 4A). Cluster 1 contained 96 samples, of which 74 were the advanced-stage samples and 22 the early-stage samples. Cluster 2 contained 35 samples, of which 18 were the advanced-stage samples and 17 were the early-stage samples. The overall accuracy of classification was 69.47% (91/131), similar to the results obtained for training dataset. Chi-square test showed that the distribution of early and advanced samples were significantly different between the two clusters. Besides, the KM survival analysis showed that cluster 1 also had a better survival prognosis than cluster 2 (logRank p = 0.059; Fig. 4B). The SVM classifier was then used to classify samples in the validating dataset. The results showed that the SVM classifier could accurately identify 112 of 131 samples. The overall accuracy was 85% (sensitivity = 98%, specificity = 56%, PPV = 84%, NPV = 92%) and the AUROC was 0.952 (Fig. 4C), which was consistent with the results obtained for training dataset. The KM survival analysis showed that the prognosis of the early-stage-like group was significantly better than that in the advanced-stage-like group (logRank p = 0.016; Fig. 4D).
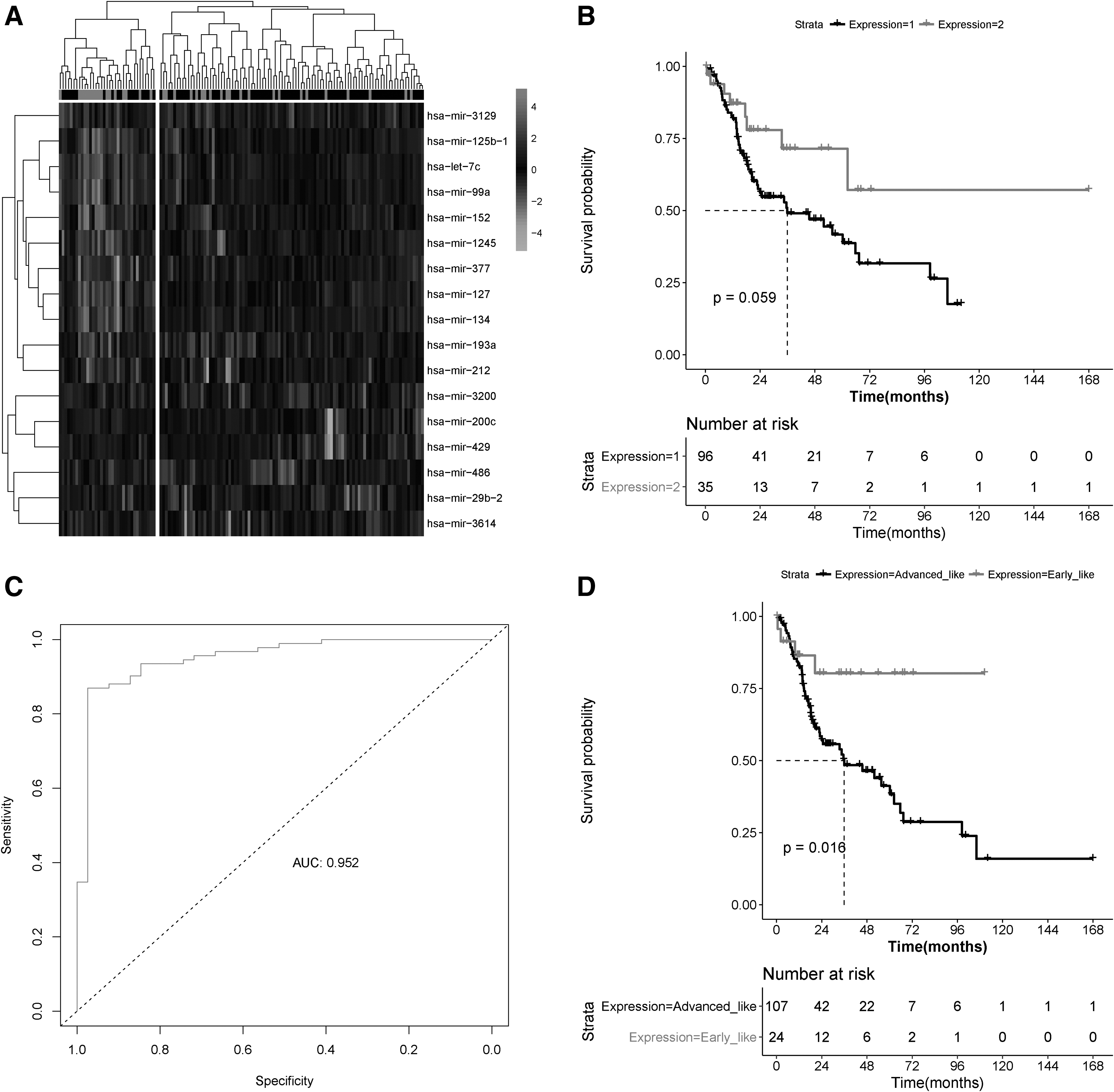
Validation of the optimal subset of feature miRNAs.
Identification of feature miRNAs related to the pathologic grades
The 17 feature miRNAs screened in TCGA database were matched on 13 mRNAs screened in GEO database, and the 13 miRNAs corresponded to 16 probes, including hsa-let-7c, hsa-miR-1245, hsa-miR-125b-1, hsa-miR-127-3p, hsa-miR-127-5p, hsa-miR-134, hsa-miR-152, hsa-miR-193a-3p, hsa-miR-193a-5p, hsa-miR-200c, hsa-miR-212, hsa-miR-29b-2, hsa-miR-377, hsa-miR-429, hsa-miR-486-3p, and hsa-miR-486-5p (Supplementary Data S1). Besides, the expression violin plot of 16 probes were drawn, and compared with the normal group; most miRNAs were tended to be upregulated or downregulated (Supplementary Data S2).
Functional annotation of feature miRNAs
It is well known that miRNA can post-transcriptionally regulate mRNA. The mRNA profile between different progressions of bladder cancer was analyzed. Compared with the early-stage group, 765 mRNAs showed significantly different expression in the advanced-stage group (99 downregulated genes and 666 upregulated genes). Target genes of the 17 feature miRNAs were predicted using TargetScan7.2 and miRDB and used to construct an miRNA-mRNA regulation network (Fig. 5A). The network contained a total of 323 nodes (14 miRNAs and 309 mRNAs) and 470 edges. Topology properties were analyzed and degree centrality, betweenness centrality, and closeness centrality scores were acquired. The nodes with scores of 0 were removed from the miRNA-mRNA network and a subnetwork was constructed using the remaining nodes. As a result, the subnetwork consisted of 128 nodes (7 upregulated and 5 downregulated miRNAs, 108 upregulated and 8 downregulated mRNAs) and 276 edges (Fig. 5B). To reveal the potential roles of the feature miRNAs in the progression of bladder cancer, enrichment analysis was performed using the mRNAs in the miRNA-mRNA subnetwork. As given in Figure 5C and D, the clusters with the highest enrichment scores were related to the extracellular matrix (ECM) and structure, collagen trimmer, and PI3K-Akt signaling pathway.

Functional annotation of feature miRNAs
Discussion
In this study, we analyzed the public data of bladder cancer from TCGA and identified 52 significantly differentially expressed miRNAs between different progression stages of bladder cancer. Among them, 17 feature miRNAs were identified and used to construct an SVM classifier, which showed high performance in discriminating early-stage from advanced-stage bladder cancer. Moreover, we constructed an miRNA-regulated target gene network and identified ECM process and PI3K-AKT signaling pathway regulated by miRNA. Findings of this study may act as important therapeutic targets or prognostic markers in progression of bladder cancer.
Numerous studies were aimed to screen the miRNAs associated with bladder cancer. For instance, Braicu et al. found that miR-139-5p, miR-143-5p, miR-141b, miR-200s, and miR-205 could be used for the prognostic biomarkers of bladder cancer. 26 Yin et al. shown that 21 miRNAs were demonstrated to be significantly related to the prognosis of bladder cancer. 27 However, the analysis of these studies were performed between the tumor and adjacent normal bladder tissue. In this study, the differentially expressed miRNAs were screened between different progression stages of bladder cancer. Moreover, among the 17 feature miRNAs, 11 showed higher expression in advanced stage than in early stage, such as miR-1245, miR-125b-1, and miR-let-7c. In primary breast tumors, miR-1245 was considered as an oncogene and it acted as a potent suppressor of the tumor suppressor protein BRCA2. 28 In natural killer cells, miR-1245 attenuated the expression of NKG2D, an activating receptor involved in tumor immunosurveillance. 29 MiR-1245 was also upregulated in lung cancer tissues. 30 On the contrary, six miRNAs were downregulated in advanced stage. For example, miR-200c inhibited invasion and migration in human colon cancer cells by targeting ZEB1. 31 Overexpression of the microRNA miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin, triggering cancer invasion and metastasis. 31 The data showed that the feature miRNA subset can be a marker for progression of the bladder cancer.
According to our functional annotation analysis, ECM-related pathway (ECM) enrichment is dominant in the progression of bladder cancer. ECM is an essential component of the tumor microenvironment. Deregulated ECM dynamics are a hallmark of cancer, which has effects on the biological characteristics of cancers. 32,33 Moreover, cancer progression is associated with increased ECM deposition and crosslink. 34 The chemical and physical signals elicited from ECM are necessary for cancer cell proliferation and invasion. 35 Moreover, our study showed that the upregulated Akt-PI3K pathway is enriched by mRNAs in the miRNA-mRNA subnetwork. Researchers have reported that aberrant activation of the PI3K/AKT pathway promotes the survival and proliferation of tumor cells in many human cancers. 36 –38 Taken together, our results showed that the feature miRNAs may play important roles in bladder cancer progression.
In this study, we constructed an SVM classifier based on the feature miRNAs. The accuracy rate of bidirectional hierarchical clustering for diagnosis of bladder cancer progression is ∼68%, whereas the SVM classifier is >85%, suggesting that SVM classifier has more accuracy for the diagnosis of progression of bladder cancer. Moreover, compared with a single molecular marker as previous studies discovered, 39 the SVM classifier is more comprehensive in the diagnosis of the progression of bladder cancer. However, we also recognized the limitations of our study. The data analyzed in this study derives from the public database and not generated by us. In addition, more validation data are needed to confirm the feature miRNAs we identified. Besides, further experimental studies are needed to provide a detailed understanding of the mechanisms related to the prognostic miRNA combination in bladder cancer. Further larger sample sizes are needed in the future to screen the miRNAs related to the progression of bladder cancer.
In conclusion, we comprehensively reanalyzed the expression profiles of bladder cancer samples and identified 17 feature miRNAs, which were significantly associated with the progression of bladder cancer. These key prognostic miRNAs may serve as promising molecular markers for diagnosis and outcome prediction of bladder cancer in future clinical practice.
Footnotes
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (No. 81372318); a grant (No. PWRL2017-07) supported by Pudong New District Commission of Health and Family Planning Leading Talent Program (Jianghua Zheng), Shanghai, China; Science and Technology Commission Fund of Shanghai Fengxian District (No. 20160907).
Supplementary Material
Supplementary Data S1
Supplementary Data S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.