
Abstract
Background:
Colorectal cancer (CRC) ranks as the third most common cancer, accounting for a significant number of cancer-related deaths worldwide every year. Yet, the molecular mechanisms responsible for the progression of this malignancy are not fully understood. Numerous studies indicate that BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B) plays a role in the progression of various malignant tumors. However, the specific biological functions and the detailed mechanisms of how BUB1B influences CRC are still not completely known. This study aimed to explore the expression and role of BUB1B in CRC.
Materials and Methods:
To achieve this, the expression levels of BUB1B in human CRC tissues and cell lines were examined using real-time polymerase chain reaction and Western blotting. The role and associated mechanisms of BUB1B in CRC cell progression were assessed both in vitro and in vivo using RNA interference.
Results:
The findings of this study revealed an elevated expression of BUB1B in both CRC tissues and cell lines. The silencing of BUB1B in CRC cell lines notably inhibited cell proliferation, migration, and invasion, leading to cell cycle arrest and apoptosis. In addition, the knockdown of BUB1B inhibited the JNK/c-Jun signaling pathway, increased the expression of proapoptotic proteins, and decreased the expression of antiapoptotic proteins. The effects of BUB1B knockdown on CRC cell progression were reversed by the JNK activator PAF(C-16).
Conclusions:
In summary, the suppression of BUB1B hindered malignant tumor progression and heightened apoptosis and cell cycle arrest in CRC cells via the JNK/c-Jun pathway. Importantly, the removal of BUB1B expression curtailed tumor growth in human CRC xenografts in nude mice, suggesting its potential as a promising therapeutic target for CRC patients.
Introduction
Colorectal cancer (CRC) stands as a dominant gastrointestinal cancer, resulting in significant global morbidity and mortality. 1 The rate of CRC occurrence is on the rise in numerous developed nations, making it the third most diagnosed cancer in America. 2 Recent findings suggest that primary prevention methods for this disease include dietary adjustments, nutritional supplements, and regular exercise. The decline in CRC mortality rates can be attributed to advances in early diagnosis, chemotherapy, biological therapy employing antibodies, immunotherapy, or a combination of these treatments. 3,4 However, the precise molecular mechanisms behind colorectal carcinogenesis remain elusive. There is a pressing need for research that identifies early CRC diagnostic markers, which will shed light on its progression mechanism and enhance therapeutic strategies.
The BUB1 mitotic checkpoint serine/threonine kinase B (BUB1B) is instrumental in the regulation of the spindle assembly checkpoint protein family. 5 This kinase is crucial for correct chromosome separation during cell division. Numerous studies suggest that elevated BUB1B expression is linked with the emergence of various tumors. For instance, a marked increase in BUB1B expression was observed in prostate cancer, which further stimulated cancer cell growth. 6 An escalated expression of BUB1B also contributed to the progression of extrahepatic cholangiocarcinoma through the JNK (c-Jun N-terminal kinase)/c-Jun signaling pathway. 7
Similarly, overexpression of BUB1B was detected in hepatocellular carcinoma, driving progression via the activation of the mTORC1 signaling pathway. 8 These findings underscore the critical role of BUB1B in tumor development. Previous research employed both bioinformatics analysis and experimental techniques to highlight BUB1Bs potential importance in CRC prevention and treatment. 9 Another study ranked BUB1B among the top 20 genes showing heightened expression in CRC samples relative to standard tissue. 10 However, the comprehensive role of BUB1B in CRC remains ambiguous.
The recent study by the authors revealed an increased expression of BUB1B in CRC tissues and cell lines. The authors identified BUB1B as a contributor to CRC tumorigenesis by influencing malignant tumor development and apoptosis in CRC cells. BUB1B achieved these effects primarily through the activation of the JNK/c-Jun pathways. In addition, CRC xenograft experiments in nude mice indicated that the suppression of BUB1B significantly curbed tumor growth. Drawing from these insights, the results of this study establish BUB1B as a promising therapeutic target for CRC.
Materials and Methods
Clinical specimens
CRC tissue samples (n = 47) and adjacent tissues were collected from Yueyang Central Hospital between July 2019 and February 2021 via surgical resection. This group comprised 27 males and 20 females with an average age of 51.2 ± 7.2 years, ranging from 41 to 71 years. Before surgery, none of the patients underwent treatments, such as radiotherapy, chemotherapy, targeted therapies, or immunobiotherapy. From the clinically extracted tumor and neighboring tissues, some fresh samples were stored in liquid nitrogen, whereas others were preserved using formalin, embedded in paraffin, and then sectioned for diagnostic purposes. Experienced pathologists confirmed the CRC diagnosis through histopathological analysis. These patients did not suffer from other severe conditions (such as stroke, coughing up blood, rectal bleeding, and heart attack), and comprehensive clinicopathological data are available. The ethics committee of Yueyang Central Hospital sanctioned this research (Approval No. 2019 K-C086), and all involved patients provided their written informed consent.
Main reagents
The JNK activator PAF(C-16) was sourced from Sigma (St. Louis, MO). Cell culture essentials, including fetal bovine serum (FBS), RPMI-1640 medium, and Dulbecco's modified Eagle's medium (DMEM), were acquired from Thermo Scientific (Grand Island, NY). The Cell Counting Kit-8 (CCK-8) was procured from Dojindo (Tokyo, Japan), whereas the Annexin V/PI apoptosis detection kit came from BD Biosciences. TRIzol reagent was sourced from Invitrogen, and propidium iodide was from KeyGen Biotech Co., Ltd. (Nanjing, China). The First Strand cDNA Synthesis Kit was obtained from Thermo Fisher Scientific, Inc. (Waltham, MA), and the quantitative reverse transcription polymerase chain reaction (qRT-PCR) reagent kit was from Takara, Inc. (Japan). The anti-BUB1B antibody (11504–2-AP) was procured from Proteintech (Wuhan, China). Antibodies such as p-JNK (sc-6254), anti-JNK (sc-7345), anti-bax (sc-7480), anti-p-c-Jun (sc-53182), anti-c-Jun (sc-74543), and anti-GAPDH (sc-47724) were purchased from Santa Cruz Biotechnology (CA). In addition, the anti-cleaved caspase-3 antibody (#9579) and anti-bcl-2 antibody (#3498) were sourced from CST (Boston).
Cell culture
The regular colon epithelial cell line (NCM460) and six CRC cell lines (LoVo, HT-29, HCT116, SW620, Caco-2, SW480) were acquired from the Chinese Academy of Science (Shanghai, China). All CRC cell lines were cultured in RPMI-1640 medium, whereas the NCM460 cells were maintained in DMEM supplemented with 10% FBS and 100 U/mL of penicillin/streptomycin. These cells were kept in an incubator with 5% CO2 at a temperature of 37°C.
Establishment of stable BUB1B knockdown cell lines
To generate stable BUB1B knockdown cell lines, CRC cell lines HT-29 and Caco-2 were seeded into 6-well culture plates at a density of 6 × 105 cells per well during their exponential growth stage. Once they reached ∼50% confluence after 24 h, these cells were transfected with lentivirus vectors: pLVX-shRNA (NC), pLVX-shRNA-BUB1B-1 (shRNA-BUB1B-1), and pLVX-shRNA-BUB1B-2 (shRNA-BUB1B-2). This was performed according to the manufacturer's guidelines using the polybrene reagent. After transfection, 0.5 μg/mL of puromycin was added to the culture medium to select successfully transfected cells, resulting in stable BUB1B knockdown cell lines. Both the BUB1B-knockdown and negative control (NC) lentivirus vectors were created and provided by Bio-Transduction Lab Co. Ltd. (Wuhan, China).
The sequences for shRNA-BUB1B-1, shRNA-BUB1B-2, and NC are as follows: shRNA-BUB1B-1: 5′-GCACAAGAATCTGCCTGTAACAATATTCAAGAGATATT GTTACAGGCAGATTCTTGTGCTTTTT-3′; shRNA-BUB1B-2: 5′-CAGACAGCTT GTGGCACTATCTACATTCAAGAGATGTAGATAGTGCCACAAGCTGTCTGTTT TT-3′; NC: 5′-GCAA AGAGTCTGTCCCAATAACATATTCAAGAGATATGTTATT GGGACAGACTCTTTGCTTTTT-3′. The stability and effectiveness of the BUB1B knockdown in these cell lines were later verified through Western blot and qRT-PCR analyses.
Determination of cell viability assay
The stably transfected cells were placed in 96-well plates and kept in an incubator at 37°C with 5% CO2. They were cultured for different time intervals: 12, 24, 48, and 72 h. After each time point, a 10 μL CCK-8 solution was added to each well and incubated for an hour, following the manufacturer's instructions. The microplate reader (VICTOR Nivo 3F; Perkin Elmer) was utilized to measure the optical absorbance at 450 nm, allowing the assessment of cell viability. Moreover, the stably transfected Caco-2 cells were exposed to PAF(C-16) at a concentration of 1 μg/mL for 24 h, whereas some cells were not exposed to it. Afterward, the CCK-8 solution was added to each well again to determine the optical absorbance. The resulting data were then used to analyze the cell viability curve.
Migration and invasion assays
In this experiment, the authors obtained stably transfected cells and subjected them to 24 h of serum starvation. After that, the authors took 100 μL of cells at a concentration of 2 × 105/mL and placed them in the upper chambers of the assay. The top chamber contained serum-free medium, while the lower chamber had medium with 20% FBS. The cells were allowed to migrate at 37°C in a humidified incubator with 5% CO2 for 24 h. Nonmigratory cells in the upper chamber were then removed, and the cells that had migrated to the lower surfaces of the membranes were quantified by staining them with 0.1% crystal violet. For the cell invasion assay, the procedure was similar, except that the Transwell membranes were pre-coated with Matrigel. After washing the invasive cells with phosphate-buffered saline (PBS) three times, they were fixed and stained with 0.1% crystal violet before observation under an upright Leica microscope. The authors counted cells in five random fields for all groups.
Flow cytometry analysis
To evaluate the rate of cell apoptosis, the cell apoptosis detection kit was used. All cell groups were collected and suspended in 300 μL of 1 × binding buffer. Each sample was then incubated with 5 μL of Annexin V-FITC in a dark room at room temperature for 10 min. After that, the cells were washed three times with 1 × binding buffer and resuspended with an additional 100 μL of 1 × binding buffer before adding 5 μL of PI. For cell cycle analysis, the cells were harvested, washed three times with PBS, and fixed overnight in precooled 70% ethanol at 4°C. The following day, the cells were washed, centrifuged at 1000 rpm, and then incubated with 5 μL of PI mixed with RNase in the dark for 30 min. After that, the cells were rinsed three times with PBS. Finally, the authors analyzed the different treatment cells using a flow cytometer (BD FACSAria). FlowJo v10.7.1 software and CellQuest software were used for the analysis.
RNA extraction and quantitative real-time polymerase chain reaction
The RNA was isolated from tissues and cells using the TRIzol reagent. After determining the RNA concentration and purity, the reverse transcription-PCR was conducted with the First Strand cDNA Synthesis Kit to obtain complementary DNA (cDNA) and qRT-PCR reagents. PCR was performed using the CFX96 Touch Real-Time PCR System (Bio-Rad). 11 The qRT-PCR conditions comprised an initial pre-denaturation at 95°C for 5 min, followed by denaturation at 95°C for 30 s, annealing at 65°C for 30 s, extension at 72°C for 30 s, and then 35 cycles of amplification.
Finally, there was a final extension step at 72°C for 10 min. To determine gene expression, the data were normalized to glyceraldehyde 3-phophate dehydrogenase (GAPDH) using the relative quantification method based on Ct values obtained from PCR products. The relative mRNA expression was calculated using the 2−ΔΔCt method. 12 Primers sequences were follows: BUB1B: forward, 5′-AAATGACCCTCTGGATGTTTGG-3′; reverse, 5′-GCATAAA CGCCCTAATTTAAGCC-3′; GAPDH: forward 5′-ATGACATCAAGAAGGTGGTG-3′; reverse: 5-CATACCAGGAAATGAGCTTG-3′.
Western blotting assay
For all the treated cells, the authors collected them and lysed them with RIPA buffer containing PMSF (Beyotime, China) on ice for 30 min. After centrifugation at 4°C and 12,000 rpm for 15 min, the protein concentration was determined using the BCA assay. The proteins were then separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 12%) and transferred onto polyvinylidene fluoride membranes with a pore size of 0.22 μm. Subsequently, the membranes were blocked using 5% bovine serum albumin (BSA) for 1 h at room temperature. Next, the membranes were incubated with primary antibodies against BUB1B (1:1000), p-JNK (1:1000), JNK (1:1000), bax (1:1000), p-c-Jun (1:1000), c-Jun (1:1000), cleaved caspase-3 (1:1000), bcl-2 (1:1000), and GAPDH (1:1000). After that, the appropriate secondary antibodies were added at a dilution of 1:10,000. The bands were visualized using the ChemiDoc Touch Gel Imaging System (Bio-Rad) through chemiluminescence. The intensity of the target protein bands was measured and analyzed using the ImageJ software.
Nude mice xenograft model
For the in vivo assay, male nude mice (∼4 weeks old and ∼12–16 g) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. The mice were randomly divided into two groups for the experiments. HT-29 cells (1 × 106 per mouse) transfected with either NC or short hairpin RNA (shRNA)-BUB1B were suspended in 100 μL of PBS and injected into the right forelimb of each nude mouse. The authors monitored the tumors every week and measured their length and width. To calculate the tumor volumes, the following formula was used: length × width 2 × 0.5. After 4 weeks, the mice were sacrificed under anesthesia.
The tumors were excised, weighed, rinsed with PBS, and then fixed with 4% paraformaldehyde. The authors stained 5 μm sections for histological examination using hematoxylin-eosin (HE) staining. All housing and procedures were carried out in accordance with the approved protocols by the Committee for the Management and Use of Laboratory Animals of Disease Control and Prevention of Hubei Province (Approval No. 202310060). The authors ensured proper care for all animals, and the study protocols were compliant with the institution's guidelines.
HE and immunohistochemical staining
Tumor tissues were obtained from euthanized animals and then preserved in 10% buffered formalin. Afterward, the tissues were embedded in paraffin blocks, sectioned, and stained with HE for examination under an optical microscope (Olympus Optical Co., Tokyo, Japan). For immunohistochemical staining, the sections underwent a 15 min treatment with citrate buffer (pH 6.2) to retrieve antigenicity. Then, the endogenous peroxidase activity was blocked with 3% H2O2 for 10 min at room temperature. Following that, the sections were blocked with 5% BSA and incubated overnight at 4°C with the appropriate primary antibodies against BUB1B (1:200), p-JNK (1:200), and p-c-Jun (1:200). After rinsing with PBS, the tissue sections were exposed to biotinylated secondary antibodies at room temperature for 1 h. The antibody complex was detected using the streptavidin–peroxidase reaction kit with DAB (Beijing Zhongshan Golden Bridge Biotechnology Ltd., Beijing, China), and finally, images were acquired and photographed using Olympus.
Apoptosis analysis by TUNEL
The tumor tissue sections were washed with PBS and then subjected to the TUNEL (KeyGen Biotech Co., Ltd.) assay, following the manufacturer's protocol for detecting apoptosis. The apoptotic cells showed localized green fluorescence, whereas the cell nuclei emitted blue fluorescence. Images were captured using an Olympus IX51 fluorescence microscope.
Statistical analysis
The experiments were repeated three times. The data analysis was carried out with SPSS 22.0 software (SPSS, Inc.). To assess the significance of data between two groups, Student's t-test was used. For comparing variables, one-way analysis of variance followed by post hoc LSD-t method was employed. Statistical significance is represented in figures by: *p < 0.05; **p < 0.01, ***p < 0.001.
Results
BUB1B is overexpressed in CRC tissues and cell lines
To examine the role of BUB1B in CRC, the authors initially analyzed its expression in CRC cancer specimens and normal controls using the Gene Expression Profiling Interactive Analysis (GEPIA) database. The results showed higher BUB1B expression in CRC tissue samples compared with normal tissue (Fig. 1A). However, no significant difference was found in BUB1B expression concerning the prognosis of CRC patients (Fig. 1B). This lack of difference might be attributed to factors, such as follow-up time, data analysis method, sample heterogeneity, and data quality, which require further verification. To delve deeper, the authors conducted qPCR to measure BUB1B mRNA levels in CRC cancer and adjacent tissues from 47 CRC patients.
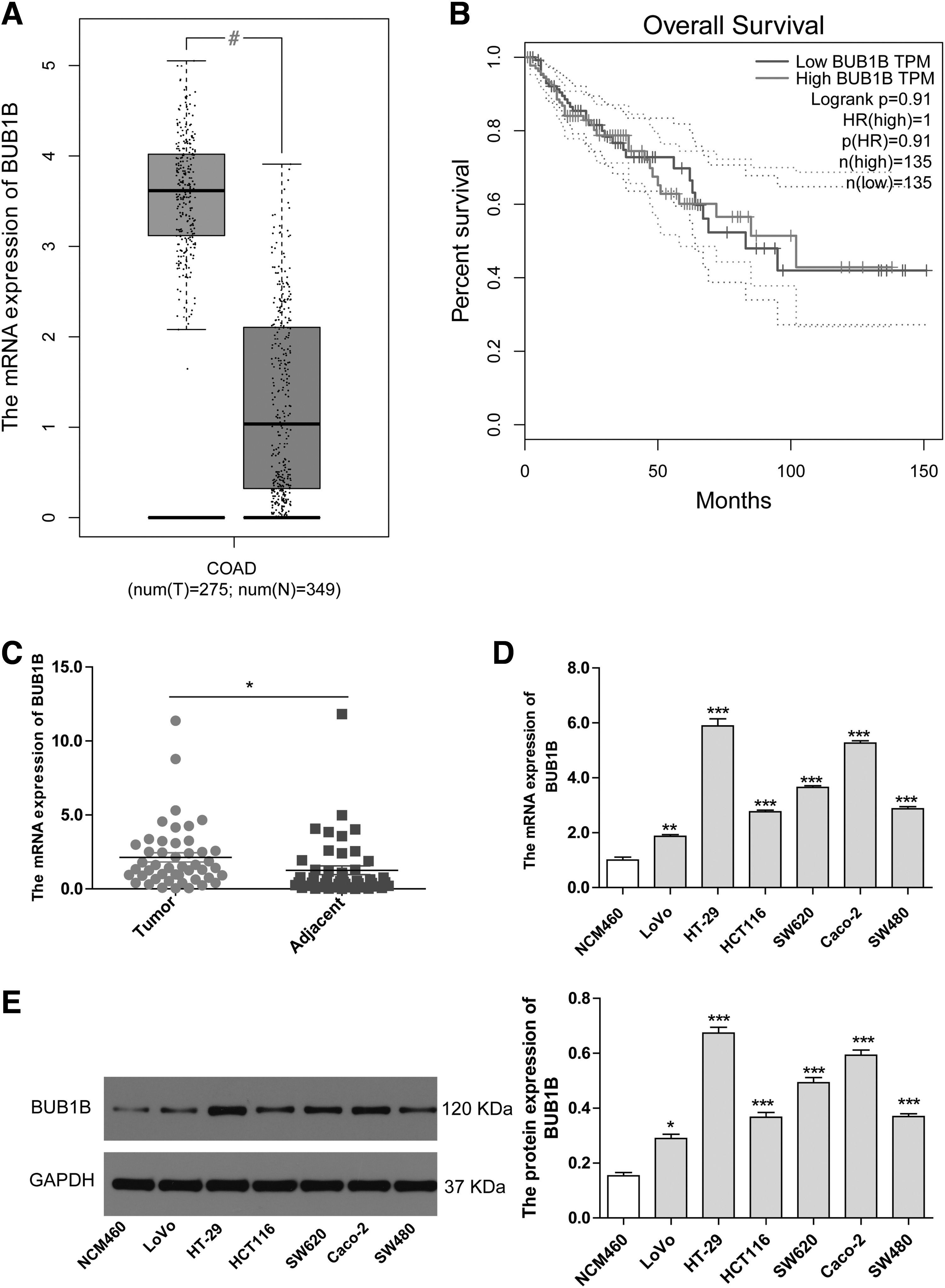
BUB1B expression is higher in CRC tissues and cell lines.
The findings revealed a significant increase in BUB1B mRNA levels in CRC cancer tissue compared with adjacent tissue (Fig. 1C). This observation was supported by the upregulation of BUB1B mRNA expression in common CRC cell lines such as LoVo, HT-29, HCT116, SW620, Caco-2, and SW480, when compared with normal colon cells (NCM460) (Fig. 1D). Furthermore, the protein expression also demonstrated the overexpression of BUB1B in these CRC cell lines (Fig. 1E). These results suggest that BUB1B is highly expressed in both CRC tissue and cell lines. Among the tested CRC cell lines, HT-29 and Caco-2 showed the highest levels of BUB1B mRNA and protein expression. Consequently, the authors chose these two cell lines for further investigations.
Knockdown of BUB1B contributes to cell apoptosis and cell cycle blocking
In this research, the authors used lentiviral vectors containing BUB1B shRNA (sh-BUB1B-1 and sh-BUB1B-2) and an NC to investigate the impact of BUB1B alteration in CRC cell lines. The authors transfected HT-29 and Caco-2 cell lines with sh-BUB1B-1, sh-BUB1B-2, and NC. The results from qRT-PCR and Western blotting showed a significant decrease in BUB1B expression in the BUB1B knockdown group compared with the NC group in both cell lines (Fig. 2A, B), with sh-BUB1B-1 demonstrating the highest interference efficiency among the two shRNA sequences. Moreover, the CCK-8 assay revealed that BUB1B knockdown substantially reduced CRC cell proliferation compared with the NC group (Fig. 2C, D).

Knockdown of BUB1B inhibits CRC cell proliferation and proapoptosis.
In addition, flow cytometry analysis confirmed that BUB1B knockdown significantly promoted apoptosis in CRC cell lines compared with the NC group (Fig. 2E, G). Furthermore, knocking down BUB1B increased the proportion of cells in the G1 phase while reducing the number of cells in the S phase in both cell lines (Fig. 2F, H). This suggests that BUB1B knockdown restricts the proliferation of CRC cells by arresting the cell cycle at the G1 phase and promoting cell apoptosis.
Knockdown of BUB1B suppresses CRC cell migration and invasion
The Transwell assays showed that reducing BUB1B significantly weakened the migration and invasion abilities of CRC cell lines compared with the NC group (Fig. 3A, B). The Western blotting results confirmed that BUB1B knockdown decreased the expression of Bcl-2, an antiapoptotic protein, while enhancing the expression of pro-caspase-3 and Bax, which are proapoptotic proteins (Fig. 3C). Previous studies have highlighted the importance of the JNK pathway in cell proliferation, apoptosis, migration, and invasion. 13 In this study, when BUB1B was knocked down, the expression of p-JNK and p-c-Jun was reduced (Fig. 3D). This suggests that BUB1B mediates the malignant behavior of CRC cells through the JNK signaling pathway.
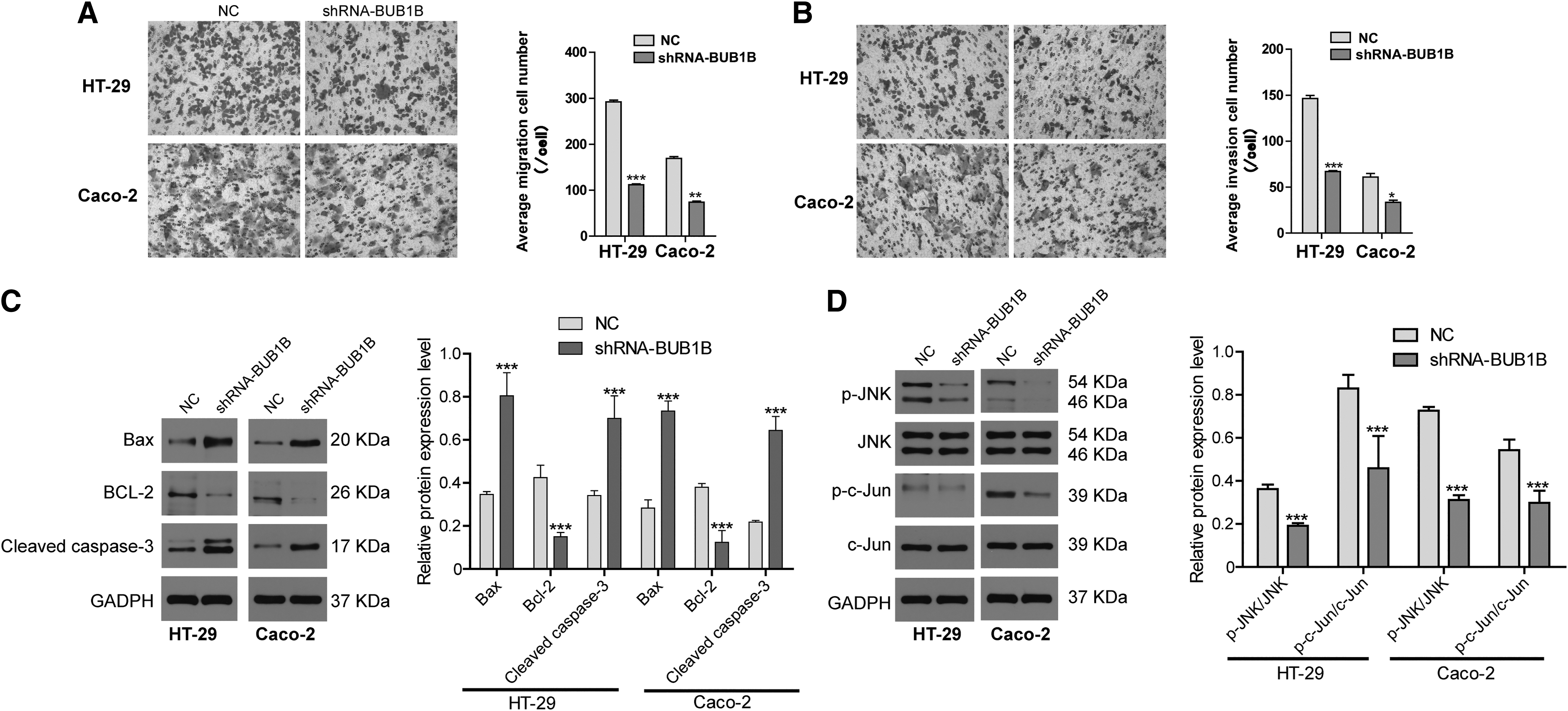
Knockdown of BUB1B inhibits CRC cell migration and invasion through JNK/c-Jun signaling.
Activation of JNK inhibits cell apoptosis and promotes cell migration and invasion in BUB1B-knockout CRC cells
PAF(C16) is a potent activator of the MAPK and MEK/ERK pathways, 14,15 with the JNK pathway being downstream of the MAPK signaling pathway. In this study, the authors used PAF(C16) to test the effects of BUB1B knockdown on CRC cell lines and its impact on cell viability, apoptosis, cell cycle, migration, and invasion. First, the CCK-8 assay showed that BUB1B knockdown reduced cell viability compared with the NC group. In addition, treating the NC group with PAF(C16) for 24 h increased the cell viability. However, when BUB1B was knocked down and PAF(C16) was added, it weakened the cell viability compared with the NC plus PAF(C16) group (Fig. 4A). This suggests that the activated JNK-c-Jun signaling pathway partially reversed the effects of BUB1B knockdown on cell viability. Second, the levels of p-JNK and p-c-Jun were significantly reduced in the sh-BUB1B group compared with the NC group. Treating the NC group with PAF(C16) increased the expression of p-JNK and p-c-Jun compared with the NC group.
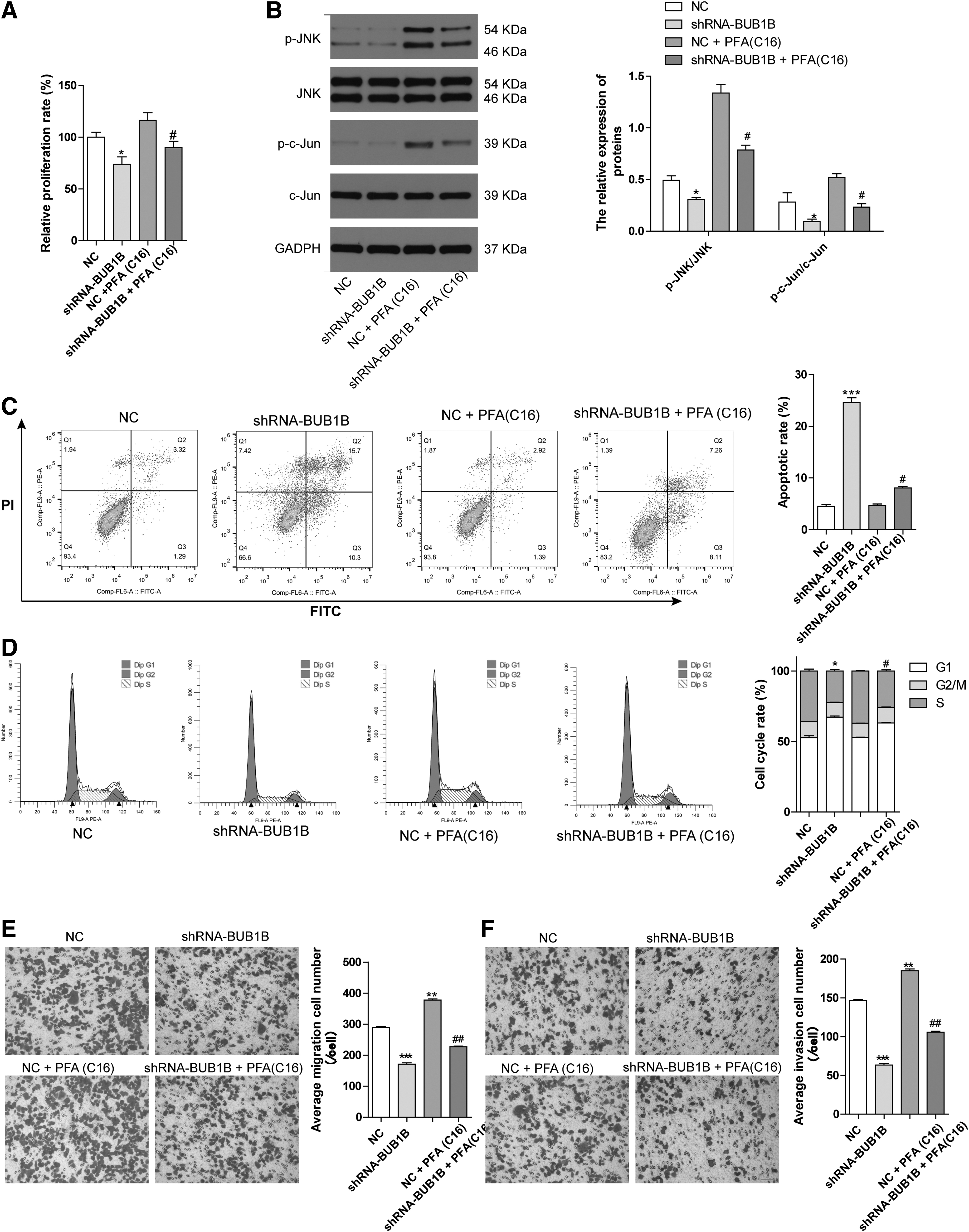
Activation of JNK/c-Jun reverses the BUB1B knockdown effects in CRC cell lines.
Furthermore, knocking down BUB1B in the group supplemented with PAF(C16) decreased the expression of p-JNK and p-c-Jun compared with the [NC plus PAF(C16)] group. However, the expression of JNK and c-Jun showed no significant changes in all groups (Fig. 4B). Third, flow cytometry analysis revealed that BUB1B knockdown significantly promoted CRC cell apoptosis and G1 phase blocking compared with the NC group. However, in the [NC plus PAF(C16)] group, the cell cycle alteration and apoptosis rate showed no significant change compared with the NC group.
When BUB1B was knocked down in the group supplemented with PAF(C16), it further promoted cell apoptosis and G1 phase blocking compared with the [NC plus PAF(C16)] group (Fig. 4C, D). Finally, the cell migration and invasion assays demonstrated that BUB1B knockdown or [NC plus PAF(C16)] reduced or promoted CRC cell migration and invasion ability compared with the NC group, respectively. Knocking down BUB1B in the group supplemented with PAF(C16) reduced the migration and invasion ability compared with the [NC plus PAF(C16)] group (Fig. 4E, F). In conclusion, BUB1B mediates CRC cell apoptosis, proliferation, migration, and invasion through the JNK-Jun signaling pathway.
Knockdown of BUB1B blocked tumor growth in xenograft nude mice model
To further explore the functional impact of BUB1B on tumor growth, the authors transfected CRC cells with either the NC or shRNA BUB1B and then injected them subcutaneously into the right forelimb of nude mice. After 4 weeks, mice implanted with shRNA BUB1B cells showed smaller tumors compared with those with NC cells, as evident from reduced tumor volume and weight (Fig. 5A–D). When the authors stained the tumor cells using HE staining, the nuclei and their fragments exhibited a blue-violet hue, whereas the cytoplasm appeared pink-red. Control mice displayed typical tumor tissue patterns with a blue-violet color (Fig. 5E). Immunohistochemistry data revealed that BUB1B knockdown downregulated the expression of BUB1B, p-JNK, and p-c-Jun, leading to increased tumor cell apoptosis (Fig. 5F, G). These results aligned with the in vitro findings of this study, indicating that BUB1B can inhibit tumor cell growth and induce cell apoptosis in vivo by modulating JNK-c-Jun signaling.
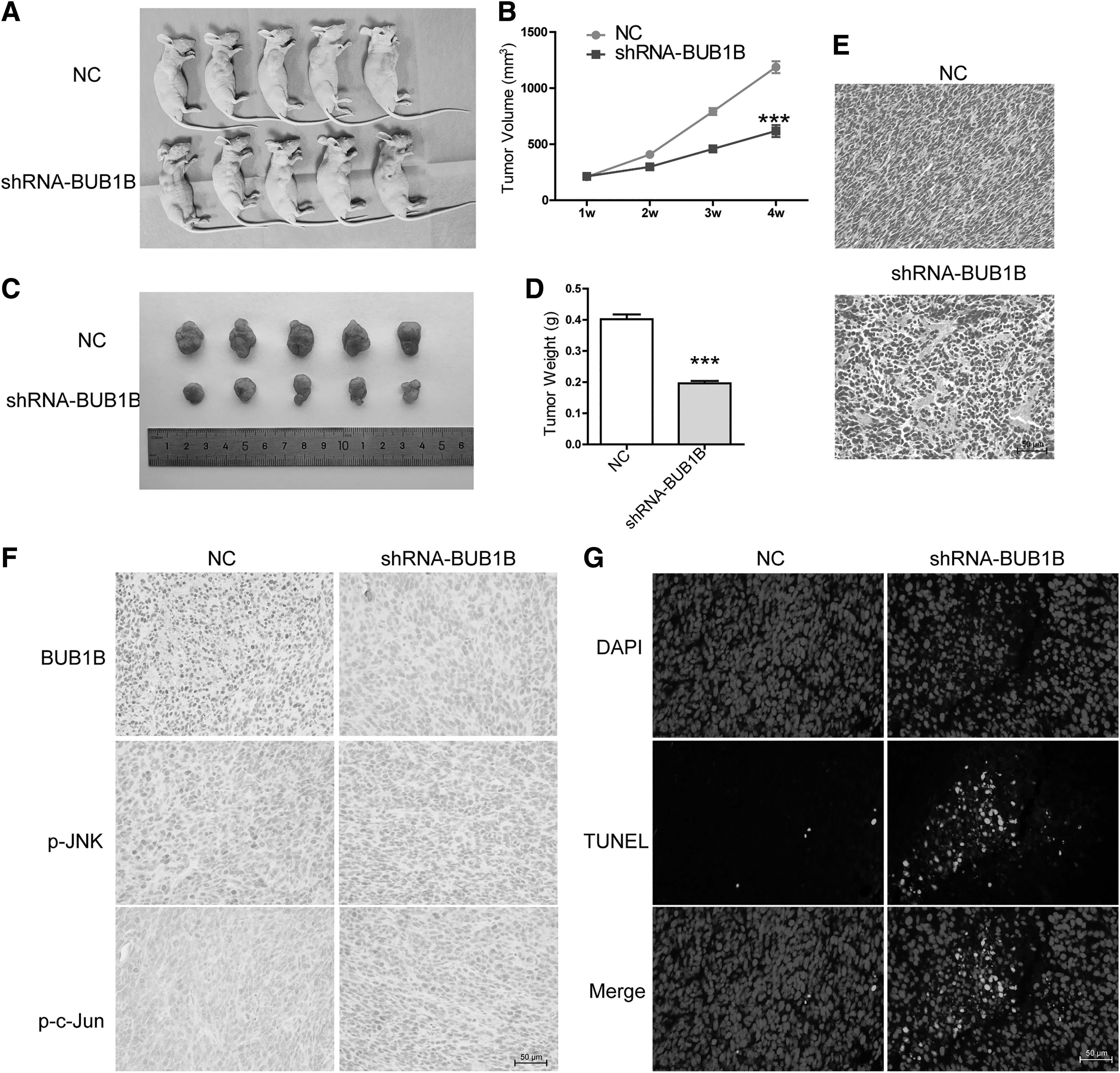
Knockdown of BUB1B blocked tumor growth in xenograft nude mouse model.
Discussion
In recent years, BUB1B has attracted increasing attention due to its involvement in various cancers, including prostate cancer, 16 cholangiocarcinoma, 7 ovarian cancer, 17 multiple myeloma, 18 and hepatocellular carcinoma. 18 BUB1B shows promise as a potential target for inhibiting tumor progression and serving as an anticancer therapy against different human cancers. Furthermore, bioinformatic analyses have indicated higher BUB1B expression and its association with the development of CRC. 9,10 However, the biological function of BUB1B in CRC remains unclear. In this study, the authors observed significantly higher BUB1B expression in CRC tissues. Knocking down BUB1B effectively inhibited the malignant progression of CRC cells, promoting cell apoptosis and cell cycle arrest. Mechanistically, BUB1B activated the JNK-c-Jun signaling pathway, contributing to tumorigenesis both in vitro and in vivo.
Tumor cells migrate and invade in response to specific external signals, such as chemical and mechanical signals. Hence, blocking tumor cells' migration and invasion could be a promising approach for cancer treatment. This study demonstrated a significant reduction in cell proliferation, migration, and invasion upon BUB1B knockdown in CRC cells. Similarly, knocking down BUB1B in cholangiocarcinoma cells showed decreased proliferation and invasiveness in both in vitro and in vivo settings. 7 Conversely, overexpression of BUB1B had opposite effects. Furthermore, knocking down BUB1B also hindered the malignant progression of thyroid carcinoma cells. 19 These collective findings suggest that suppressing BUB1B expression can effectively impede tumor cell migration and invasion.
Importantly, a study on prostate cancer cells showed that overexpressing BUB1B promoted cell proliferation, migration, and invasion, while depleting BUB1B did not affect these cellular functions. 20 Since the current study did not involve BUB1B overexpression, future research should explore its role in upregulating proliferation, migration, and invasion in CRC cells. Tumorigenesis involves multiple signaling pathways, including JNKs, which belong to the MAPK protein family. JNK signaling regulates various biological processes such as inflammation, cell proliferation, migration, survival, and cell death. 21 It also controls the Bcl family of proteins (Bcl-2, Bcl-XL, Bim, and Bad) to mediate cell apoptosis. Blocking the JNK pathway inhibits cell proliferation and promotes apoptosis. 22,23
In this investigation, the authors observed that inhibiting BUB1B led to the suppression of the JNK-c-Jun signaling pathway, upregulation of proapoptotic proteins such as Bax and cleaved caspase-3, and downregulation of the antiapoptotic protein Bcl-2. To further confirm the role of BUB1B in regulating JNK signaling in CRC cells with BUB1B knockdown, the JNK signaling activator PAF(C16) was used. Interestingly, the effects of BUB1B knockdown in CRC cells were reversed after supplementing with PAF(C16). This indicates that BUB1B mediates the JNK-c-Jun signaling, influencing CRC apoptosis. Similar roles of BUB1B have been reported in breast cancer and thyroid carcinoma. 19,24 In a study on thyroid carcinoma cells, BUB1B knockdown disrupted cell cycle progression, although the specific phase alterations were not mentioned. 19
In this study, the authors found that BUB1B knockdown arrested the cell cycle at the G1 phase. Moreover, in a xenograft hepatocellular carcinoma tumor model, BUB1B knockdown significantly reduced tumor growth and metastasis. 8 Similarly, reducing BUB1B expression suppressed malignant tumor progression and decreased tumor weight and volume in a cholangiocarcinoma xenograft tumor model in mice. 7 These data also revealed that BUB1B knockdown inhibited CRC progression in vivo, providing a novel understanding of how BUB1B contributes to tumor progression. Overall, this study demonstrates that BUB1B promotes tumor growth in CRC by positively regulating the JNK/c-Jun signaling pathway.
Various methods employed in this study investigated the role of BUB1B in CRC cell lines. BUB1B was found to control the progression of malignant tumors in CRC. However, certain methods, such as BUB1B overexpression analysis, were not conducted to confirm its function in the mentioned processes. Further exploration is needed to understand the involvement of BUB1B in other cell death processes such as autophagy, 25 ferroptosis, 12,26 or cuproptosis. 27 In addition, apart from the JNK-c-Jun signaling pathway, the participation of BUB1B in other signaling pathways and its role in different types of tumors require investigation. More studies are necessary to validate the findings, and the exact molecular mechanism needs to be clarified.
In conclusion, CRC tissues displayed elevated expression of BUB1B, which was experimentally verified in multiple CRC cell lines. When BUB1B was suppressed, it hindered CRC cell proliferation, migration, and invasion while promoting cell apoptosis and blocking the cell cycle. Experimental assays also demonstrated that BUB1B regulated CRC ability through the JNK/c-Jun signaling pathway both in vitro and in vivo. Thus, BUB1B shows potential as a promising therapeutic target for CRC treatment.
Footnotes
Authors' Contributions
Q.Z. and S.Z. performed the experiments and wrote the article. L.H., Q.F., and L.L. performed the experiments. L.C. revised the article. X.D. designed the study and reviewed the article. All authors read and approved the final article as submitted.
Disclosure Statement
No competing financial interests exist.
Funding Information
This work was financially supported by the Scientific Research Project of Hunan Provincial Health Commission (No. 202203034641).