
Abstract
Background:
Natural killer (NK) cells are characterized by their antitumor efficacy without previous sensitization, which have attracted attention in tumor immunotherapy. The heterogeneity of osteosarcoma (OS) has hindered therapeutic application of NK cell-based immunotherapy. The authors aimed to construct a novel NK cell-based signature to identify certain OS patients more responsive to immunotherapy.
Materials and Methods:
A total of eight publicly available datasets derived from patients with OS were enrolled in this study. Single-cell RNA sequencing data obtained from the Gene Expression Omnibus (GEO) database were analyzed to screen NK cell marker genes. Least Absolute Shrinkage and Selection Operator (LASSO) Cox regression analysis was used to construct an NK cell-based prognostic signature in the TARGET-OS dataset. The differences in immune cell infiltration, immune system-related metagenes, and immunotherapy response were evaluated among risk subgroups. Furthermore, this prognostic signature was experimentally validated by reverse transcription-quantitative real-time PCR (RT-qPCR).
Results:
With differentially expressed NK cell marker genes screened out, a five-gene NK cell-based prognostic signature was constructed. The prognostic predictive accuracy of the signature was validated through internal clinical subgroups and external GEO datasets. Low-risk OS patients contained higher abundances of infiltrated immune cells, especially CD8 T cells and naive CD4 T cells, indicating that T cell exhaustion states were present in the high-risk OS patients. As indicated from correlation analysis, immune system-related metagenes displayed a negative correlation with risk scores, suggesting the existence of immunosuppressive microenvironment in OS. In addition, based on responses to immune checkpoint inhibitor therapy in two immunotherapy datasets, the signature helped predict the response of OS patients to anti-programmed cell death protein 1 (PD-1) or anti-programmed cell death ligand 1 (PD-L1) therapy. RT-qPCR results demonstrated the roughly consistent relationship of these five gene expressions with predicting outcomes.
Conclusions:
The NK cell-based signature is likely to be available for the survival prediction and the evaluation of immunotherapy response of OS patients, which may shed light on subsequent immunotherapy choices for OS patients. In addition, the authors revealed a potential link between immunosuppressive microenvironment and OS.
Introduction
Osteosarcoma (OS) represents the predominant subtype of primary bone sarcoma among children, adolescent, and young adult. With resection and adjuvant chemotherapy, the 5-year survival rate for nonmetastatic pediatric and young adult patients with OS has augmented to 78%, but there remains ∼20% for patients with metastasis at diagnosis or recurrence. 1 Recently, immunotherapy represented by immune checkpoint inhibitor (ICI) has attracted attention from clinicians and researchers due to its improved efficacy compared with traditional strategies. The advent of ICI has accelerated the research on immunotherapy and has indicated a potential treatment paradigm for OS. 2
ICIs are deployed to amplify the antitumor effects of immune cells. However, the prognosis of their intervention in OS is not satisfactory. 3 From the analysis of the results of numerous clinical trials, the role of ICIs to treat OS is quite limited. Only a portion of OS patients have experienced long-term benefits from immunotherapy. 4 Consequently, a relatively comprehensive understanding of the molecular mechanisms defining the OS immune competence is required for developing novel prediction models and identifying novel predictive biomarkers.
The OS microenvironment has been inextricably implicated in tumorigenesis, encompassing immune cells, bone, stromal, and vascular. 5 The tumor microenvironment (TME) represents a complex and dynamic milieu with synergistic functions in the tumor progression and metastatic dissemination. Tumor infiltration of immune cells controls tumor progression and efficacy of immunotherapy. 6 Natural killer (NK) cells, known as innate lymphoid cells with cytotoxic properties, play a crucial role in tumor immunotherapy. The cytotoxicity of NK cells is precisely governed by a complex interplay of activating and inhibitory signals mediated by their receptors.
NK cells participate in antitumor immunity by interacting directly with tumor cells or secreting cytokines and chemokines in the regulation of adaptive immunity, thus controlling the tumor growth. The importance of NK cells in mediating antimetastatic effect has been established in experimental murine tumor models. 7 Besides, analysis of 33 different types of cancer from The Cancer Genome Atlas database reveals that most of cancer tissues with marked NK cell infiltration are also infiltrated with CD8+ T cells. NK cells are capable of eliminating cancer cells that escape CD8+ T cell immunosurveillance. 8 In addition, high density of tumor-infiltrating NK cells confers a favorable clinical outcome in multiple human solid tumors. 9 While recent advances in genetic modification of NK cells have caused renewed interest in their potential for OS immunotherapies, little is known about the molecular characteristics of NK cells in OS.
The extensive application of single-cell RNA sequencing (scRNA-seq) and molecular profiling allows the systematic interrogation of tumor-infiltrating immune cells at single-cell resolution, leading to important insights into the biological characteristics of tumor-associated NK cells in the TME. 10 High-dimensional data of immune subpopulations from scRNA-seq could be used to predict patients' prognosis and immunotherapy response. 11 In this study, the authors first focused on the comprehensive analysis of scRNA-seq of OS to construct NK cell marker genes signature (NKCMGS). Then, the authors corroborated the prognostic value of NKCMGS through validation in four independent datasets from the Gene Expression Omnibus (GEO) database. The authors further analyzed the correlation of NKCMGS with the response to immunotherapy in OS.
Materials and Methods
Data collection
A total of eight public datasets of primary OS patients were retrospectively analyzed, including one scRNA-seq dataset from the GEO database, one RNA-seq dataset from the TARGET database, four independent microarray datasets from the GEO database, one anti-programmed cell death protein 1 (PD-1) immunotherapy dataset from the GEO database, and one anti-programmed cell death ligand 1 (PD-L1) immunotherapy dataset from the IMvigor210 R package. scRNA-seq data from six OS tissues of GSE162454 were utilized to screen NK cell marker genes of OS. The transcriptome and clinical phenotypic data of 89 TARGET-OS patients were utilized for screening prognosis-related genes and constructing the risk signature.
The expression profiles and clinical data from GSE16091 (n = 34), GSE21257 (n = 53), GSE39055 (n = 36), and GSE39058 (n = 41) datasets were utilized for external validation. The expression profiles and clinical parameters from GSE78220 (28 OS patients received anti-PD-1 treatment) and IMvigor210 (348 OS patients received anti-PD-L1 treatment) datasets were selected for exploring the predictive capacity of NKCMGS in immunotherapy response. These two immunotherapy datasets displayed distinct responses, including complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD). Ethical approval and informed consent were obtained in original research.
Screening of NK cell marker genes by scRNA-seq analysis
Seurat and SingleR packages were used to conduct the analysis of scRNA-seq data. The PercentageFeatureSet function was utilized to compute the proportion of mitochondrial genes to ensure that each gene was expressed in no less than five cells, expression of genes in each cell ranged between 300 and 6000, and mitochondrial gene content was <25%. The qualifying datasets underwent normalization using the LogNormalize algorithm, followed by identification of the highly variable genes (top 1500) through the FindVariableFeatures function. According to the ElbowPlot function, the first 15 principal components were subjected to t-distributed stochastic neighbor embedding analysis for dimension reduction. To obtain the differentially expressed genes among each cluster, the FindAllMarkers function was employed, and their cell types were annotated based on HumanPrimaryCellAtlasData. To identify NK cell marker genes, a cutoff threshold was applied, whereby values of p-value.adj <0.01 and |log2 FC| > 1 were considered significant.
Construction and validation of NK cell-associated prognostic signature
Genes related to prognosis from NK cell marker genes were screened in the TARGET-OS dataset using univariate Cox regression analysis. Next, prognostic correlated genes were processed with Least Absolute Shrinkage and Selection Operator (LASSO) Cox regression using the glmnet package. Tenfold cross-validation was employed to select the minimal penalty term (λ). Subsequently, with results obtained from stepwise multivariate Cox regression analysis, a novel NK cell-associated prognostic signature was constructed for the OS patients. Following calculation of the median risk score, patients were stratified into either low- or high-risk subgroup. The Kaplan–Meier (K-M) survival curves were depicted for survival analysis using the survminer package, and the predictive accuracy of the prognostic signature was evaluated using time-dependent receiver operating characteristic (ROC) curves through the timeROC package. Ultimately, the prognostic value of the signature was validated through external GEO datasets.
Functional and genomic enrichment analysis
To automate the process of Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, the clusterProfiler package was utilized to offer enrichGO and enrichKEGG of gene clusters for functional and genomic enrichment analysis, respectively.
TME score estimation and tumor-infiltrating immune cell analysis
Based on RNA-sequencing data of the TARGET-OS dataset, the ESTIMATE algorithm with the estimate package was applied for calculating TME score, including stromal score, immune score, ESTIMATE score, and tumor purity score. Normalized gene expression of the TARGET-OS dataset was adopted for the quantification of the proportions of 22 infiltrating immune cell subsets into either low- or high-risk groups using the CIBERSORT algorithm.
Gene set variation analysis
Gene set variation analysis (GSVA) can transform the gene expression matrix originating from the TARGET-OS dataset into GSVA scores of gene sets, which devote the degree of absolute enrichment of gene sets in each sample. The GSVA package was implemented to obtain the GSVA scores of seven metagene sets (HCK, IgG, Interferon, LCK, MHC-I, MHC-II, and STAT1) that had complex relationships with different immune cells and tumor clinical parameters. 12 Pearson's correlation analysis was carried out to investigate the intercorrelations among metagenes of immune-related activities and the risk score of NKCMGS. The presentation of heat map plots was achieved using the ComplexHeatmap package.
Assessment of anti-PD-1/PD-L1 immunotherapy response
Using RNA-sequencing data from the TARGET-OS dataset, PD-L1 expression was applied to evaluate the sensitivity of immunotherapy. After that, to evaluate patients' response to ICI therapy, transcriptomic data and immunotherapy response were collected from two independent immunotherapeutic datasets.
Cell cultures
The human OS cell lines U2OS and HOS and the human osteoblasts (OB) cell line were obtained from Sigma–Aldrich LLC (Shanghai, China). All the cell lines used were authenticated, and the results of mycoplasma test were negative. The OS cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, USA) containing 10% fetal bovine serum (Gibco) at 37°C and 5% CO2.The OB cell line was cultured in DMEM/F-12 medium (Gibco) at 35°C and 5% CO2.
Reverse transcription-quantitative real-time PCR analysis
Total RNA was obtained from the cells using TRIzol reagent (Invitrogen, USA), and cDNA was obtained through reverse transcription using a cDNA synthesis kit (Takara, Japan). TB Green Fast qPCR Mix (Takara) was used for reverse transcription-quantitative real-time PCR (RT-qPCR) to quantify the transcripts. The primer sequences are shown in Supplementary Table S1.
Statistical analysis
Data processing and generation of plots were performed through scripting language R software (version 4.2.1). Pearson's correlation analysis was exploited to calculate correlation coefficient between continuous variables. The Wilcoxon rank sum test was performed between the low- and high-risk subgroups. Data were analyzed for the dependence between gene expression and clinical characteristics using the Kruskal–Wallis test and chi-squared test. The K-M statistic and logarithmic sequence test was implemented for survival analysis. Statistical comparisons between OS cell lines and OB cell line were performed using Student's t-test. Corresponding p < 0.05 was indicative of statistical significance.
Ethics approval and consent to participate
The ethical approval was obtained from the Medical Ethics Committee of China Three Gorges University. Informed consent was obtained from all participants.
Results
Clustering of NK cell-associated cluster and identification of NK cell marker genes
Following quality control and normalization of scRNA-seq data, there were 41,548 cells from 6 OS samples selected for subsequent analysis (Fig. 1A). Top 1500 hypervariable gene expression profiles were subjected to principal component analysis dimensionality reduction, and then, cells were classified into 18 clusters (Fig. 1B). Subsequently, the distribution of each cell cluster was annotated according to the marker genes from the HumanPrimaryCellAtlas, of which cluster 1 was classified as NK cell cluster based on the expression of known marker genes of NK cells (Fig. 1C, D). Compared with all other clusters, 146 differentially expressed genes were generated within cluster 1 through adjusted p after Bonferroni correction and identified as OS-related NK cell marker genes. GO and KEGG enrichment analyses revealed NK cell marker genes to be mainly enriched in immune-related features, primarily in the regulation of T cell activation, external side of plasma membrane, signaling adaptor activity, and human T cell leukemia virus one infection (Supplementary Fig. S1).
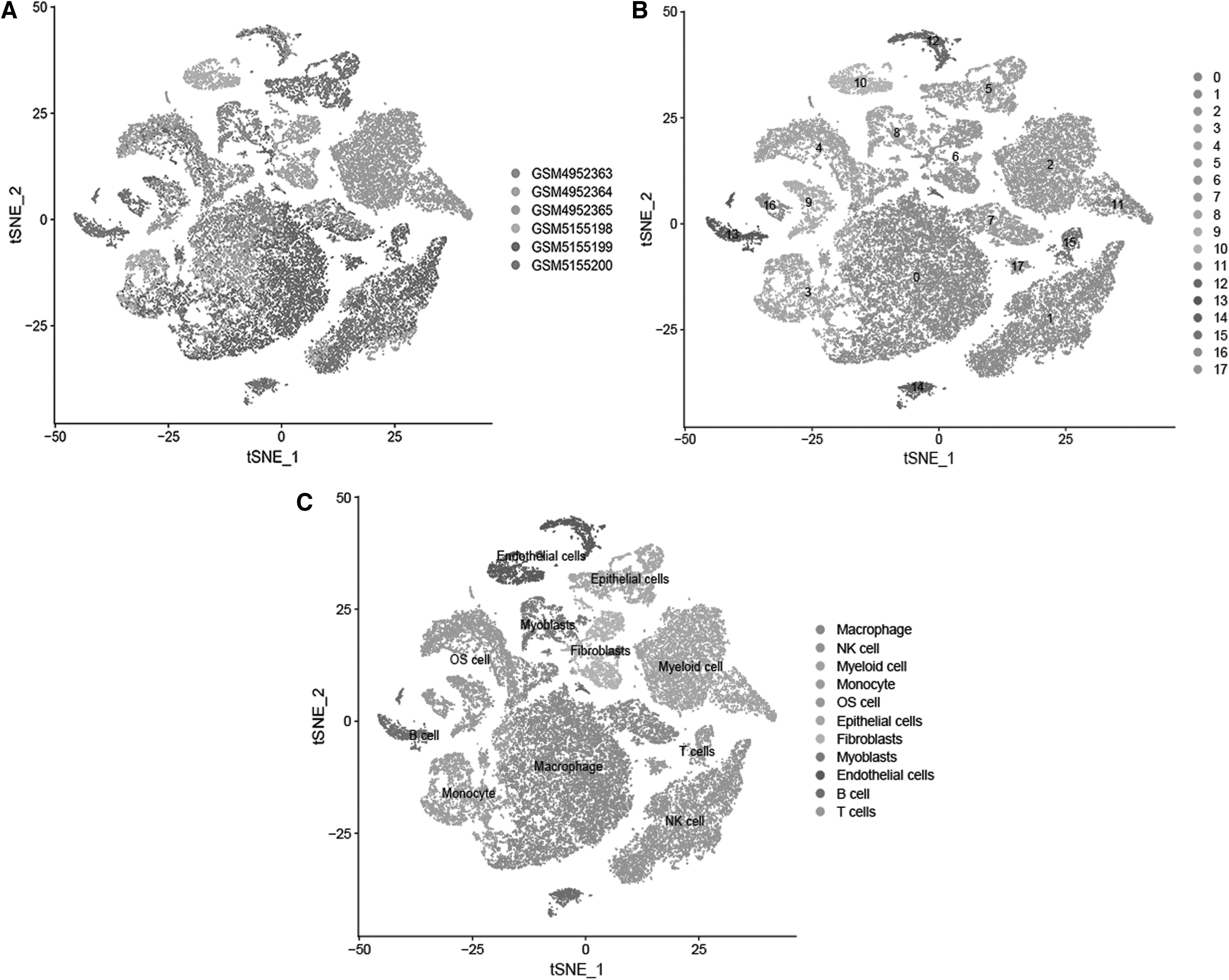
Screening of natural killer cell marker genes in single-cell RNA sequencing analysis.
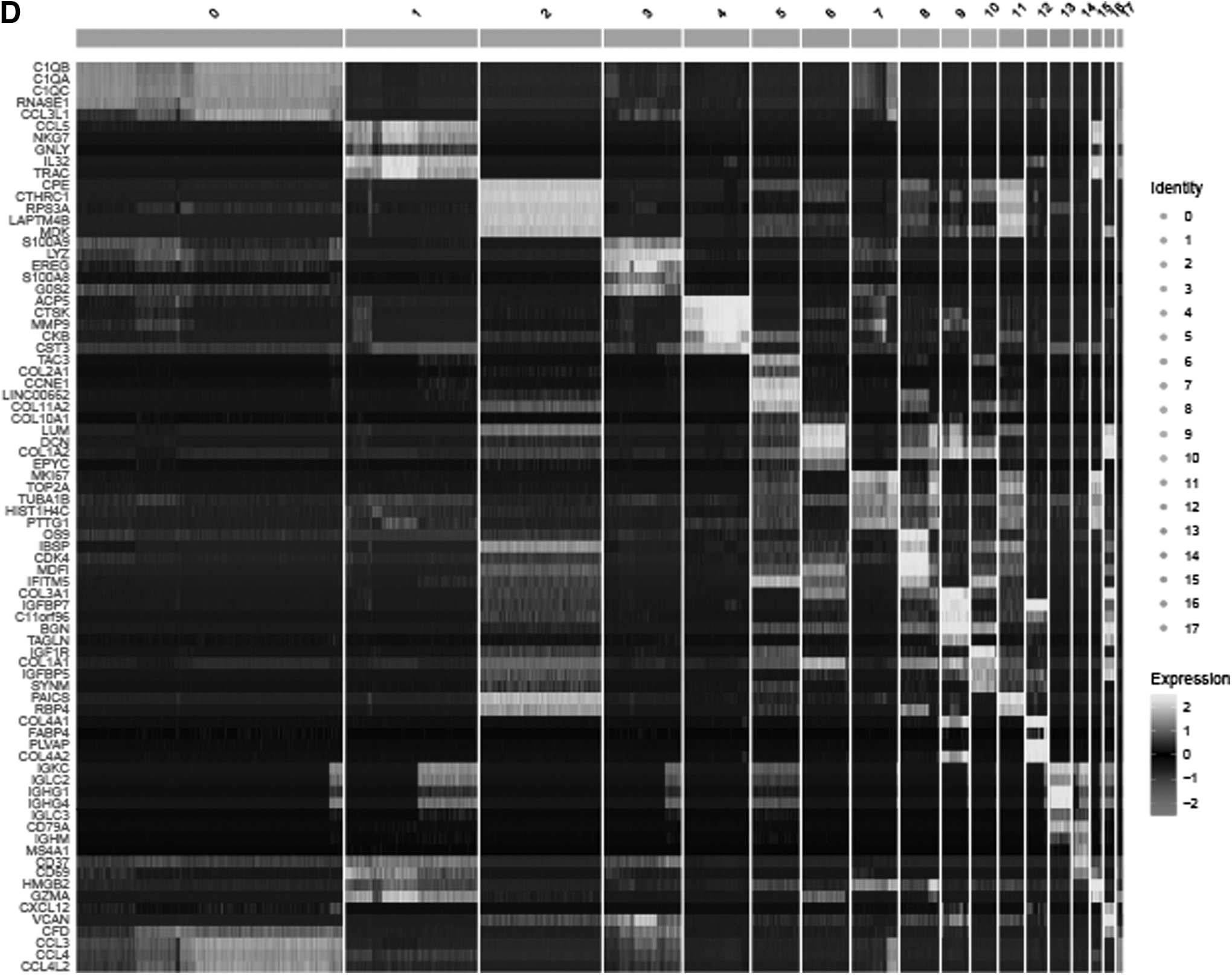
Construction of the five-gene NK cell-associated prognostic signature
The TARGET-OS dataset was used for univariate Cox regression analysis of acquired 146 NK cell marker genes, with 12 NK cell marker genes related to OS survival screened. Then, to facilitate subsequent construction of the risk signature, nine NK cell marker genes (PTPRC, ARHGDIB, LAG3, RPS29, CD37, CRIP1, DDX24, SOCS1, and RNF19A) were preserved by performing LASSO Cox regression analysis (Supplementary Fig. S2A, B). Ultimately, the optimal risk signature was constructed using stepwise multivariate Cox regression analysis by integrating five NK cell marker genes: Risk score = (−0.2562) × PTPRC + (−0.3618) × CD37 + (0.3935) × CRIP1 + (0.861) × DDX24 + (0.8651) × RNF19A (Supplementary Fig. S2C).
The expression level of the 5 genes across 18 clusters delineates the specificity of 5-gene expression in the risk signature (Supplementary Fig. S2D). According to the median risk score across all samples (median risk score = −2.779), patients in the TARGET-OS dataset were categorized into either low-risk (n = 44) or high-risk (n = 45) subgroups. Based on the scatterplots of risk scores and survival status, there was a noticeable increase in the mortality rate of OS patients as the risk score increased (Fig. 2A). The heat map displayed the mRNA expressions of five NK cell marker genes from the risk signature (Fig. 2B).

Construction of the NKCMGS in the TARGET-OS dataset.
Afterward, the K-M survival analyses demonstrated a worse clinical outcome of OS patients with high-risk score in contrast with those with low-risk score with statistical significance (Fig. 2C). The area under the curve values of the risk signature were 0.682, 0.834, and 0.883 at 1, 3, and 5 years, respectively, suggesting that it has a satisfactory predictive accuracy in the prognosis of OS (Fig. 2D). RT-qPCR results released that CRIP1, DDX24, and RNF19A were obviously elevated in U2OS and HOS cells compared with OB cell, whereas downregulation of PTPRC was observed in U2OS and HOS cells, and CD37 was only downregulated in U2OS cell (Fig. 2E). Taken together, a novel five-gene signature based on NK cell marker genes was successfully constructed.
Internal and external validation of NKCMGS in OS
Initially, to determine the robustness of NKCMGS in clinical subgroups, the TARGET-OS dataset was employed to calculate the predictive value of NKCMGS in terms of different genders and tumor staging. The subgroup analysis demonstrated that high-risk scores were significantly related to worse unfavorable survival rates in the female (p = 0.00289; Supplementary Fig. S3A) or male (p = 0.00045; Supplementary Fig. S3B), nonmetastatic (p = 5.9784e−05; Supplementary Fig. S3C) or metastatic (p = 0.04112; Supplementary Fig. S3D) OS patients. Next, four independent GEO datasets were selected to validate the predictive accuracy of NKCMGS. Patients from GEO datasets were allocated into the low- and high-risk subgroups based on the median risk score in the TARGET-OS dataset. Survival differences between the low- and high-risk subgroups in GEO datasets indicated conclusions similar to that in the clinical subgroups of the TARGET-OS dataset.
High-risk OS patients were prone to suffer from inferior prognosis, namely GSE16091 (Supplementary Fig. S4A; hazard ratio [HR] = 0.331, 95% confidence interval [CI] = 0.124–0.881, p = 0.04232), GSE21257 (Supplementary Fig. S4B; HR = 0.377, 95% CI = 0.162–0.875, p = 0.04258), GSE39055 (Supplementary Fig. S4C; HR = 0.208, 95% CI = 0.073–0.597, p = 0.02173), and GSE39058 (Supplementary Fig. S4D; HR = 0.237, 95% CI = 0.089–0.625, p = 0.03436). Similarity of ROC curves for the four validation datasets reflected that the NKCMGS can sufficiently predict 1-, 3-, and 5-year survival rates of OS patients (Supplementary Fig. S3E–H). Finally, standard meta-analysis for these four validation datasets was conducted with the Meta package for the evaluation of the integrated predictive effectiveness of NKCMGS in OS patients. Results of meta-analysis proved NKCMGS to be a robust and reliable prognostic signature for OS patients (HR = 0.289, 95% CI = 0.179–0.466, p = 3.417e−07) (Supplementary Fig. S4E).
Clinical prognostic value of the NKCMGS
To further evaluate the independent prognostic value of NKCMGS for OS patients, Cox regression analysis was conducted between the risk signature and common clinical features, including age, gender, metastasis, surgery, and chemotherapy. Univariate Cox regression analysis indicated that only risk score (HR = 4.502, 95% CI = 2.425–8.355, p = 1.852e−06) demonstrated independent prognostic significance for OS patients. After multivariate Cox regression analysis, metastasis (HR = 8.385, 95% CI = 1.738–40.440, p = 0.008), chemotherapy (HR = 11.087, 95% CI = 1.301–94.450, p = 0.027), and risk score (HR = 21.368, 95% CI = 4.657–98.029, p = 8.166e−05) were significantly related to the prognosis of OS (Table 1). These results further suggested that the NKCMGS was a promising signature for predicting the prognosis of OS.
Univariate and Multivariate Cox Regression Analyses of Natural Killer Cell Marker Genes Signature in the TARGET-OS Dataset
CI, confidence interval.
Enrichment analysis of NKCMGS-associated genes
To further explore the potential mechanisms of independent predictive capability of NKCMGS, functional enrichment analysis was employed to annotate NKCMGS-associated genes. Correlation analysis of the risk score from NKCMGS and gene expression profiles of the TARGET-OS dataset was performed to filter NKCMGS-associated genes (Pearson's |R| > 0.4, p < 0.05), with 98 positively correlated genes and 128 negatively correlated genes acquired (Fig. 3A). Afterward, GO and KEGG enrichment analyses were conducted for these genes. The majority of the enriched biological processes were related to immunological features, namely leukocyte cell–cell adhesion, regulation of leukocyte cell–cell adhesion, and immunoglobulin-mediated immune response (Fig. 3B). The KEGG pathways enriched in immune-related infection were the significant pathways related to NKCMGS-associated genes (Fig. 3C).

Functional enrichment analysis of NKCMGS-related genes.
Immune cell infiltration status of NKCMGS in TME
Considering the important role of NK cell-dependent antitumor immunity, the relationship between the NKCMGS and immune infiltrating profile in OS patients requires attention. Initially, compared with the high-risk group, the low-risk group had significantly higher TME scores (except tumor purity, including stromal score, immune score, ESTIMATE score) through the ESTIMATE algorithm analysis (Fig. 4A–D), implying a negative association of risk score and the infiltrating level of immune cells. Subsequently, the CIBERSORT algorithm was employed for the evaluation of the infiltration abundance of immune cells in TME.
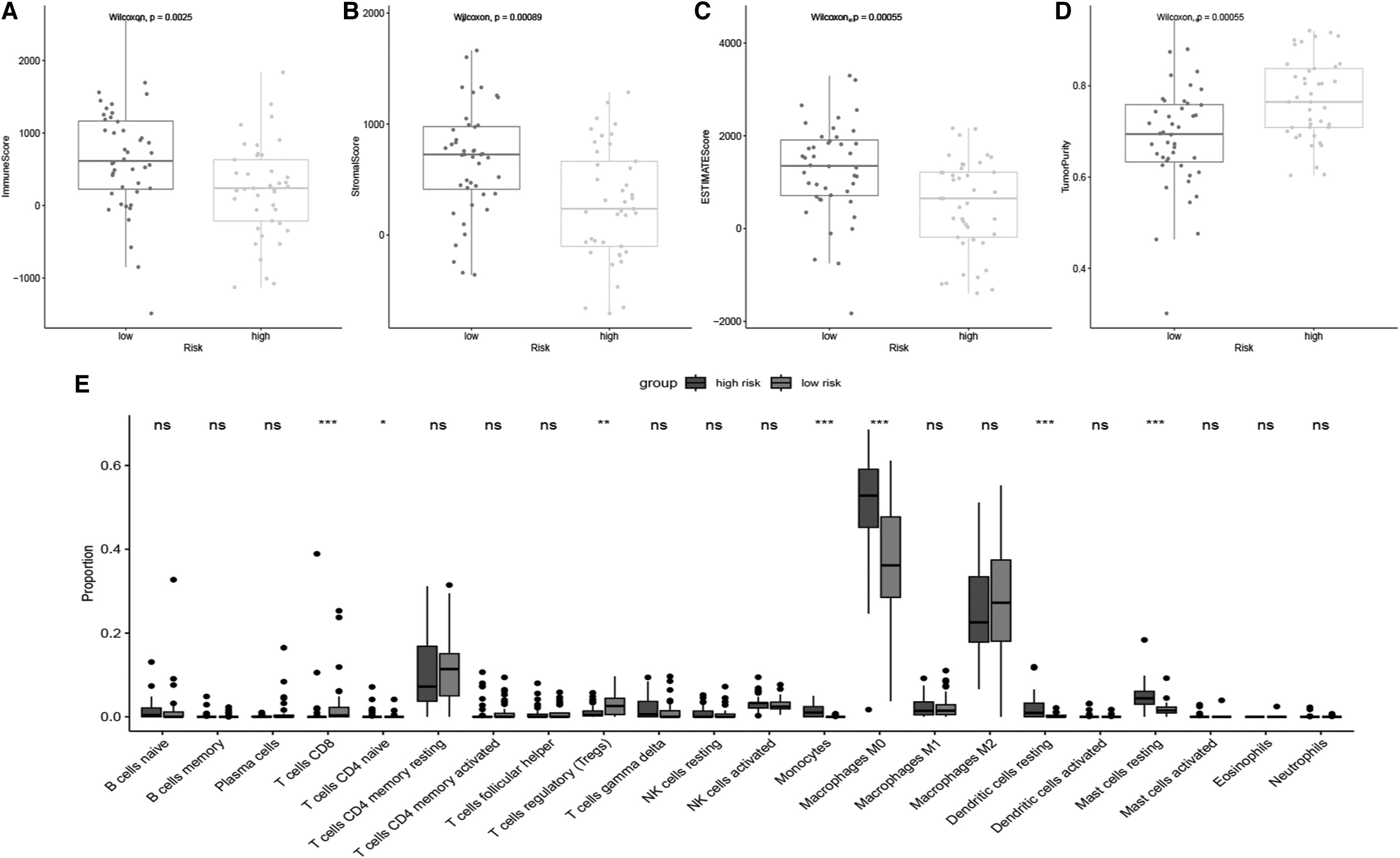
Correlation of NKCMGS with immune cell infiltration in the TARGET-OS dataset. TME-related scores of the low- and high-risk groups in the NKCMGS, including immune score
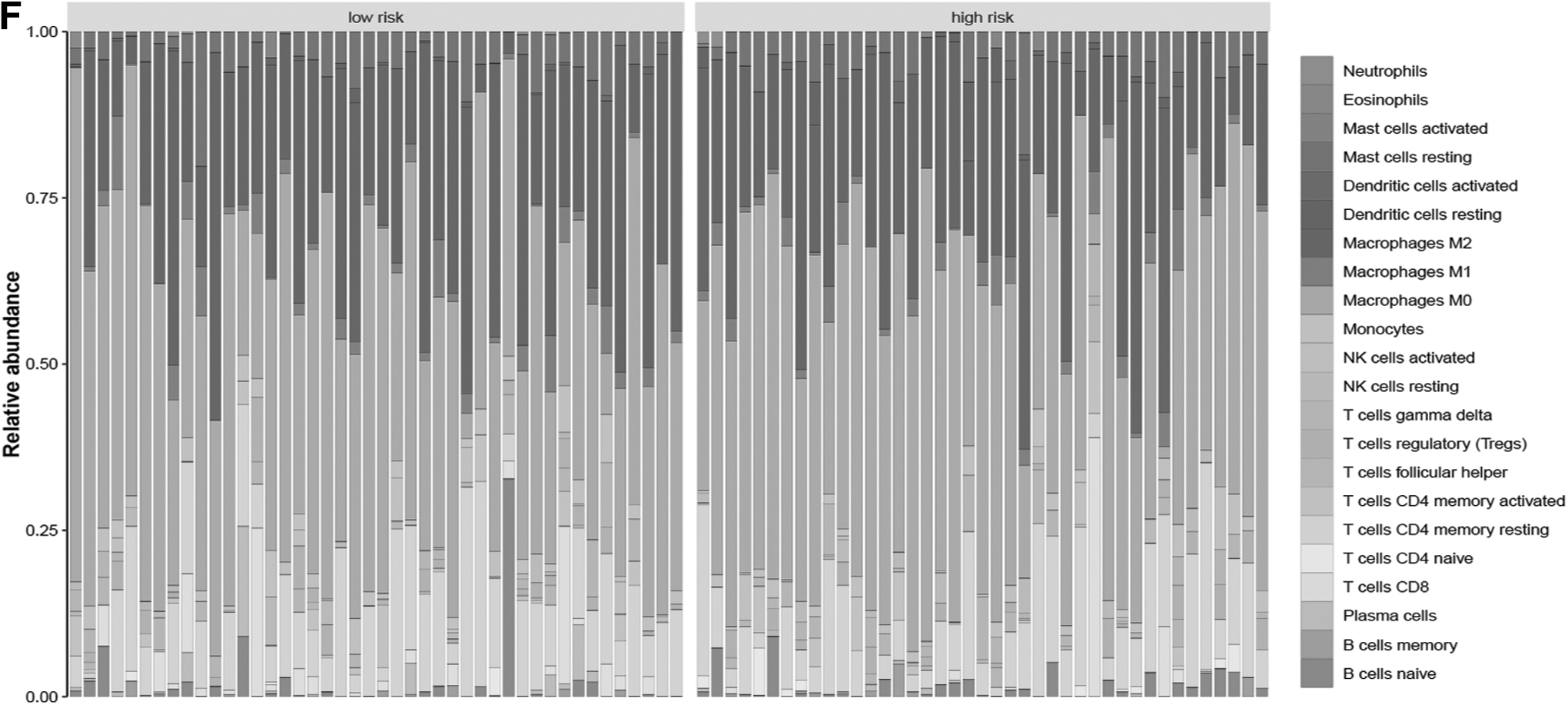
In accordance with the infiltration data from the CIBERSORT algorithm, the low-risk group exhibited higher abundance of CD8 T cells, naive CD4 T cells, and regulatory T cells, whereas the high-risk group exhibited higher abundance of monocytes, M0 macrophages, resting dendritic cells, and resting mast cells (Fig. 4E). Relative percentage of infiltrating immune cells in each risk group is shown in Figure 4F. Furthermore, correlation analysis was conducted on risk score and immune cell infiltration data and revealed a positive association of risk score with M0 macrophages and resting dendritic cells, but a negative association of risk score with plasma cells (Supplementary Fig. S5).
Immune system-related metagenes function of NKCMGS
To elucidate the associations of NKCMGS with immune system-related metagene profiles, Pearson's correlation analysis was conducted on NKCMGS and seven clusters of metagenes (HCK, IgG, Interferon, LCK, MHC-I, MHC-II, and STAT1) that had a positive correlated prognostic value in tumor subtypes by acting as surrogate markers of different immunological cell types. These metagene expressions in the TARGET-OS dataset are shown in the heat map in Figure 5A. GSVA was used to calculate the GSVA score of each cluster of metagenes, and the correlation of the risk score of NKCMGS with the GSVA score of seven clusters of metagenes was investigated. As shown in Figure 5B, IgG, Interferon, LCK, MHC-I, MHC-II, and STAT1 were identified to be negatively related to risk score.
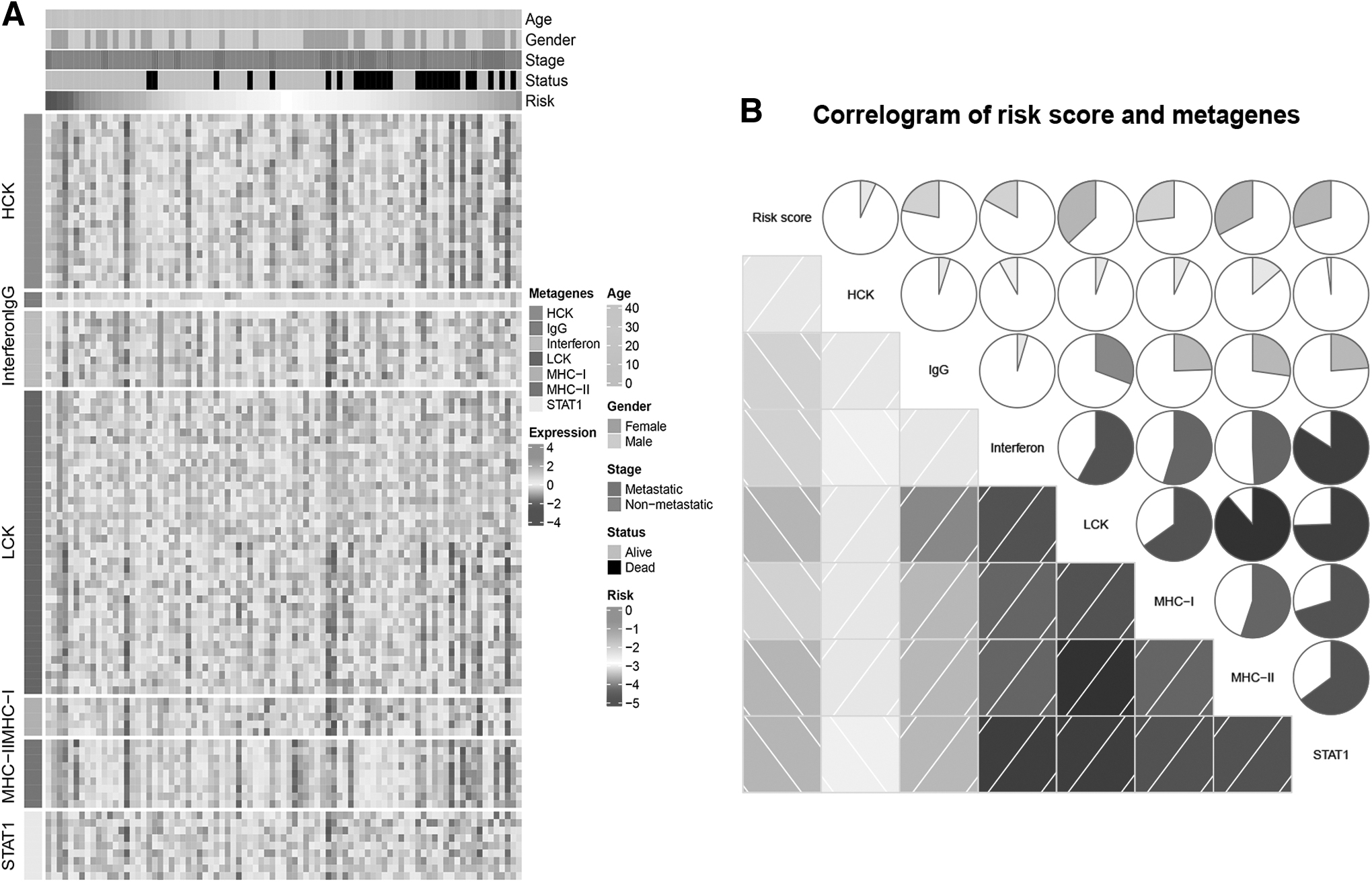
Correlation analysis between the NKCMGS and immune system-related metagenes in the TARGET-OS dataset.
Prediction of immunotherapy benefits based on NKCMGS
Given the exponentially growth of NK cell-based antitumor therapy and the increasing enthusiasm for developing the potential of NK cell-mediated immunotherapy, two immunotherapy datasets were used to explore whether NKCMGS could predict ICI immunotherapeutic response for OS patients. Initially, the relationship of widely known immune checkpoint protein (PD-L1 expression) and NKCMGS was determined in the TARGET-OS dataset. Results demonstrated a higher PD-L1 expression in the high-risk group compared with the low-risk group (Fig. 6A). The following content focused on the efficient predictive value of NKCMGS in the ICI therapy. In the anti-PD-1 dataset, patients responding to SD/PD showed higher risk score compared with patients responding to CR/PR (Fig. 6B).

Evaluation of immunotherapy benefits based on NKCMGS.
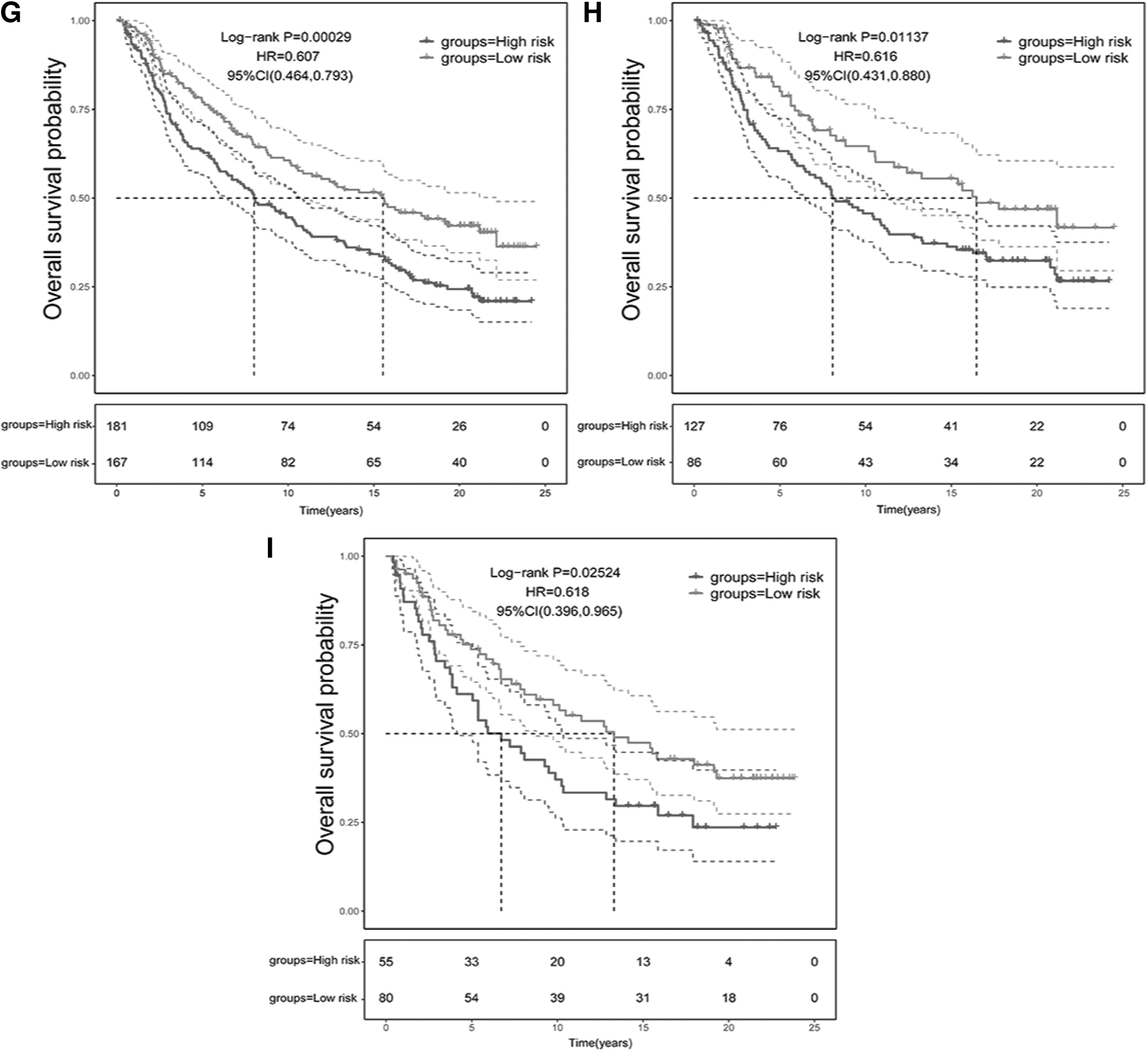
The response rate of anti-PD-1 treatment remarkably increased in the low-risk group (Fig. 6C). In addition, the K-M analysis revealed significant survival advantages for the low-risk group after anti-PD-1 treatment (Fig. 6D). In the anti-PD-L1 dataset, the low risk score was linked with CR/PR to the treatment (Fig. 6E). The proportion of CR/PR patients in the low-risk group was significantly higher in contrast with that in the high-risk group (Fig. 6F). Besides, the high-risk score in the IMvigor210 dataset was related to a relatively poorer prognosis (Fig. 6G). Similar outcomes were also observed in both early and advanced patients of this dataset (Fig. 6H, I). Overall, the analysis of these results suggested that the NKCMGS had a satisfactory predictive potential in immunotherapy efficacy and may identify low-risk OS patients who were likely to benefit more from ICI immunotherapy.
Discussion
Comprehensive profiling of tumor-infiltrating immune cells in TME is critical for realizing immune cell modulation in tumor progression and providing guidance for the development of tumor immunotherapies. The advent of scRNA-seq has fueled the explosion of molecular characteristics of infiltrating immune cells in TME. Although massive efforts have been devoted to adaptive immune cell characterizations, innate immunity has also gradually gained traction in recent years. NK cell, as a major component of the innate immune system, was a specialized immune effector cell type in antitumor immunity. Fast-acting feature of NK cells against tumor cells and overall safety of NK cell infusion have largely spurred considerable efforts for developing NK cell-based immunotherapies. 13
Thus, understanding the NK cell characteristics in the TME of OS facilitates us to develop novel predictive signatures of immunotherapy response and to improve the responsive rate. In the present study, the authors attempted to screen the NK cell marker genes of OS by scRNA-seq analysis. Based on the filtered NK cell marker genes, the authors constructed a novel five-gene NK cell-associated prognostic signature for OS patients in the TARGET database. Its prognostic accuracy was verified in four external datasets from the GEO database. Immune infiltrate analysis revealed that the low-risk score of NKCMGS was characterized by abundant infiltrating immune cells in TME. In addition, OS patients with low-risk score were closely correlated with higher response rate to immunotherapy, suggesting that ICI therapy may be efficacious for low-risk OS patients.
Reviewing the five NK cell marker genes (PTPRC, CD37, CRIP1, DDX24, RNF19A) in the NKCMGS, the majority of them are related to the mechanism in tumorigenesis or the tumor prognosis. PTPRC, also known as CD45, is a transmembrane receptor-type protein tyrosine phosphatase, fulfilling a regulating role in the innate immunity. 14 PTPRC regulates the innate immunity signaling through modulating lymphocyte survival and cytokine responses; thus, altered PTPRC could result in immunodeficiency or increased susceptibility to malignancy. 15 Overexpression of PTPRC is related to the good prognosis in small cell lung cancer patients. 16 CD37 is an extensively expressed tetraspanin protein mediating immune regulation and tumor suppression. 17 Deficiency of CD37 drives tumor development and is linked to unfavorable overall survival. 18
CRIP1, belonging to the LIM/double zinc-finger protein family, is aberrantly expressed in various solid tumors. CRIP1 promotes tumor cell migration and invasion via the Wnt/β-catenin signaling pathway in cervical cancer, and CRIP1 overexpression is connected with inferior survival. 19 DDX24 is a family member of the DEAD-box RNA helicases, which participates in tumor initiation and progression through modulating tumor cell proliferation, metastasis, and apoptosis. The elevated DDX24 expression level in non-small cell lung cancer (NSCLC) is also correlated with a poor prognosis. 20 RNF19A is a ring between ring fingers family E3 ligases that have been recognized as mediators linking to genomic alterations and malignant phenotypes. 21 RNF19A promotes tumor cell proliferation, inhibits apoptosis in NSCLC, and exerts oncogenic effects by modulating p53 ubiquitination. RNF19A overexpression is associated with poor outcome. 22 Explorations focusing on genes identified in the NKCMGS will help pave the way to provide alternative potential targets in OS treatment.
The accuracy and feasibility of NKCMGS in prognostic prediction for OS patients have been validated in both internal and external datasets, making the understanding of the underlying mechanisms behind superior predictive capability of NKCMGS to be crucial. The authors first obtained 226 NKCMGS-associated genes through correlation analysis, and GO and KEGG analyses found these correlated genes to be dominantly involved in the immune-associated regulations or immune-related infections. Thus, the inferior prognosis of OS patients may be partly derived from immune cell dysfunction, contributing to the dysfunctional state of TME. Mounting evidence reveals that tumor-infiltrating immune cells are part of TME that provides support for tumor growth and development. Limited infiltration of intratumoral immune cells imposes barriers to antitumor immunity. 23,24
The authors then calculated the TME scores and the abundance of infiltrating immune cells in the low- and high-risk groups. Results showed that TME scores and immune cell infiltration were significantly decreased in the high-risk group, suggesting the suppressed antitumor immunity. In addition, higher infiltration of CD8 T cells and naive CD4 T cells in the low-risk group but higher infiltration of M0 macrophages in the high-risk group implied that immune exhaustion states may emerge in the high-risk OS patients. Effective antitumor immunity requires the presence, activation, and co-stimulation of CD8 T and CD4 T cells. 25 CD8 T cells exert a core influence on the mediation of antitumor immunity, and intratumoral CD4 T cells are also capable of exhibiting cytotoxic activity against tumor cells. 26,27
Thus, T cell exhaustion originating from the immunosuppressive mechanisms of the TME can be used by tumors to evade immune surveillance. 28 Increasing proportions of M0 macrophages infiltration is linked to poor prognosis. 29 M0 macrophage polarization and tumor-associated macrophages infiltration contribute to tumorigenesis and metastases through immune suppression. 30 Hence, the low level of immune cell infiltration and immunosuppressive TME may exist in solid OS, possibly partly accounting for the low survival rates of high-risk OS patients.
Meanwhile, the authors further evaluated the associations between the NKCMGS and immune system-related metagene profiles, which demonstrated that the risk score seems to have a negative concordance in most clusters of metagenes. HCK mainly regulates the proliferation, migration, and secretion function of macrophages, promoting an immunosuppressive TME. 31 LCK is expressed in all T lineage cells, regulating T cell activation, T cell development, and T cell homeostasis, potentiating T cell immunotherapeutic responses. 32 MHC-I and MHC-II primarily deliver tumor antigens to CD8 T cells and CD4 T cells, whose role in antitumor immune response is receiving increasing attention. 33
STAT1 enables CD4 T cells survival by maintaining sufficient MHC-I expression, and the activated STAT1 exerts tumor-suppressive function. 34,35 Therefore, the correlation of these clusters of metagenes with different immune cell types seems to be able to evaluate the immune cell infiltration and immunosuppressive TME in tumor, implying different prognostic values of metagene expression. 12,36 In brief, the above findings indicate that underlying mechanisms behind predictive capability of NKCMGS may stem from immune cell dysfunction and immunosuppressive TME.
The discrepancy of prognosis and TME immune cell infiltration based on NKCMGS facilitates us to investigate the sensitivity of NKCMGS in predicting immunotherapy response of OS patients. Initially, the authors explored the association of widely known immune checkpoint protein (PD-L1 expression) and NKCMGS. The results revealed that PD-L1 was upregulated in the high-risk OS patients, indicating the potential differences of the immune function between the low- and high-risk OS patients. PD-L1 on tumor cells binds to its receptor PD-1, leading to tumor immunosuppression and immune escape, which explains the tendency of PD-L1 expression to be associated with poor clinical prognosis. 37 Nonetheless, it seems controversial that high PD-L1 expression usually indicates better efficacy in ICI therapy. Expression of PD-L1 in TME was not predictive in all immunotherapy response studies. 38
The clinical success of tumor ICI therapy is more linked to the extent and functionality of the T cell infiltration. 39 In this study, the low-risk OS patients exhibited higher infiltrating levels of CD8 T cells and naive CD4 T cells. Therefore, the authors then validated the predictive value of NKCMGS in two ICI therapy datasets (anti-PD-1 in the GSE78220 dataset and anti-PD-L1 in the IMvigor210 dataset). In the two immunotherapy cohorts, the authors tested the ability of NKCMGS on immunotherapeutic benefit prediction and noted that OS patients with low-risk score to anti-PD-1 or anti-PD-L1 were always sensitivity to ICI therapy, pointing to the extent and functionality of tumor-infiltrating T cells as important in immunotherapy response. Together, low-risk OS patients achieved better therapeutic benefits following ICI therapy, which more importantly confirmed the immunotherapeutic benefit prediction of NKCMGS.
Despite novel insights into immune-related therapy of OS obtained, several limitations need to be illustrated in the present study. First, the NKCMGS was established relying on NK cell marker genes from scRNA-seq data, but the spatial heterogeneity of immune microenvironment varies temporally along with tumor evolution. Therefore, the predictive capability of NKCMGS for prognostic and therapeutics effectiveness is limited. Second, there exists somewhat heterogeneity in data processing and patient population among different datasets. Therefore, the clinical validations of NKCMGS in the prospective trials involving OS patients are required for further studies. Third, the mechanistic analysis behind the predicting feasibility of NKCMGS originates from bioinformatics, and further functional researches are necessitated for validation of its biological mechanisms.
In conclusion, the authors aim to develop a five-gene prognostic signature based on a panel of five NK cell marker genes, which provided a potential predictor for the prognosis and ICI response of OS patients. The present study might facilitate clinical decision-making for patient selection in ICI therapy and provides clues to explore biological mechanisms underlying immune dysfunction based on NKCMGS through the attempt to target NK cell marker genes to augment antitumor immunity.
Data Availability Statement
The datasets generated and analyzed during this study are available in the GEO, TARGET database, and IMvigor210 R package.
Footnotes
Acknowledgments
The authors would like to thank the researchers and staff of the UCSC Xena data portal, GEO databases, and IMvigor210 dataset for providing data on OS.
Authors' Contributions
Q.L. and Y.Z. conceived and designed the study. Q.L. and X.H. analyzed the datasets and prepared the figures and tables. Q.L. contributed to the writing of the article. Y.Z. reviewed the article. All authors had final consent for publication.
Disclosure Statement
There are no existing financial conflicts.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1