
Abstract
Background:
Radiolabeled antibody fragments present a promising opportunity as theranostic agents, offering distinct advantages over whole antibodies. In this study, the authors investigate the potential of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab as a theranostic agent for precise targeting of human epidermal growth factor receptor 2 (HER2)-positive cancers. Additionally, the authors aim to quantitatively assess the binding synergism in the presence of cold trastuzumab.
Materials and Methods:
F(ab′)2-pertuzumab was prepared by pepsin digestion and conjugated with a bifunctional chelator. The immunoconjugate was radiolabeled with 177Lu and characterized by chromatography techniques. Binding parameters (affinity, specificity, and immunoreactivity) and cellular binding enhancement studies were evaluated in HER2-overexpressing and triple-negative cell lines. The in vivo enhancement in tumor uptake of the radiolabeled immunoformulation was assessed in severe combined immunodeficient (SCID) mice bearing tumors, both in the presence and absence of unlabeled trastuzumab.
Results:
The formulation of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab could be prepared in high yields and with consistent radiochemical purity, ensuring reproducibility. Comprehensive in vitro and in vivo evaluation studies confirmed high specificity and immunoreactivity of the formulation toward HER2 receptors. Binding synergism of radiolabeled pertuzumab fragments in the presence of trastuzumab to HER2 receptors was observed.
Conclusions:
The radioformulation of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab holds great promise as a targeted approach for addressing HER2-positive cancers. A potentially effective strategy to amplify therapeutic efficacy involves dual epitope targeting by combining radiolabeled pertuzumab with cold trastuzumab.
Introduction
Cancer is a leading global cause of death, with ∼10 million reported deaths of 20 million cases in 2020. 1 Among various types of cancers, breast cancer stands out as the most prevalent cancer in women. This form of cancer is often associated with amplification or overexpression of the human epidermal growth factor receptor 2 (HER2) gene, found in ∼15%–20% of breast cancer cases. 2,3 The HER2 gene encodes the HER2 protein, which is a transmembrane tyrosine kinase receptor belonging to the EGF receptor (EGFR) family. 4,5
FDA-approved, recombinant, humanized monoclonal antibodies, trastuzumab (Herceptin) and pertuzumab (Perjeta®), are used for treatment for HER2-positive breast cancer. Pertuzumab targets extracellular subdomain II of the HER2 receptor, while trastuzumab targets domain IV. Both antibodies prevent dimerization of HER2 and block downstream prosurvival pathways. Trastuzumab has significantly improved the overall survival of patients with HER2-positive early and metastatic breast cancer, becoming a standard treatment modality. 6 However, therapeutic resistance to trastuzumab has emerged in about 15%–20% of patients, leading to relapse postimmunotherapy. 7,8
Pertuzumab, with its different target (extracellular dimerization domain II), inhibits HER2 heterodimerization with HER1, HER3, and HER4. 9,10 Both pertuzumab and trastuzumab activate antibody-dependent cellular cytotoxicity (ADCC) and complement dependent cytotoxicity (CDC) pathways. 11,12 A combination therapy targeting different epitopes of the same receptor (using trastuzumab and pertuzumab) is effective against HER2-overexpressing breast cancer. However, this approach is associated with resistance to immunotherapeutic agents and adverse effects such as cardiac dysfunction. 8,13,14
Owing to the limitations associated with immunotherapy, the persistent overexpression of the HER2 receptor could also be exploited for radioimmunotherapy where radiolabeled trastuzumab and pertuzumab alone or in combination could play an important role in combating malignancies. Several preclinical and clinical studies have explored radiolabeled trastuzumab 15 –17 and pertuzumab 18,19 alone, but simultaneous targeting of both the epitopes using radiolabeled pertuzumab and cold trastuzumab has not been widely studied.
Previous in silico research indicates that trastuzumab binding makes the HER2 receptor more “plastic,” enhancing the association with pertuzumab in certain regions and leading to synergistic inhibition and improved therapeutic outcomes. 20 The authors' group has shown the binding synergism of 125I-pertuzumab and 125I-F(ab′)2-pertuzumab in the presence of trastuzumab in HER2-overexpressing cell lines. 21 Additionally, in vitro and in vivo synergism of [177Lu]Lu-DTPA-pertuzumab was explored and reported in the presence of excess trastuzumab. 22
However, using intact radiolabeled trastuzumab/pertuzumab has limitations, such as slow pharmacokinetics leading to slow elimination from blood and normal tissues, resulting in low tumor/blood (T/B) and tumor/normal tissue (T/NT) ratios. 23 –26 To overcome this, antibody fragments are being explored, which display relatively faster in vivo distribution and penetration in solid tumors. Additionally, dual epitope targeting with such vectors could improve overall targeting for theranostic applications.
The present study focuses on optimizing the formulation of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab for targeting HER2 receptors and exploring the in vivo tumor uptake synergy of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab in the presence of trastuzumab. F(ab′)2 fragments of pertuzumab were generated following a reported protocol 21 and used for the study. The immunoconjugates were radiolabeled with 177Lu using a bifunctional chelator (p-SCN-Bn-CHX-A″-DTPA).
Binding parameters (affinity, specificity, and immunoreactivity) and cellular binding enhancement studies were evaluated in HER2-overexpressing cells (SK-BR-3 and SK-OV-3) and the triple-negative cell line (MDA-MB-231). The in vivo enhancement in tumor uptake of radiolabeled immunoformulations was assessed in severe combined immunodeficient (SCID) mice bearing SK-OV-3 and SK-BR-3 tumors, both in the presence and absence of unlabeled trastuzumab.
Materials and Methods
Materials
Pertuzumab (Perjeta) was procured from Roche Diagnostics, GmbH, while trastuzumab was procured from Herclon, Genentech, Inc. Lutetium-177 (specific activity 22–25 Ci/mg) was produced at the Dhruva reactor, Bhabha Atomic Research Centre (BARC), by irradiation of the enriched 176Lu target. Bifunctional chelator 2-(4-isothiocyanatobenzyl)-diethylenetriamine pentaacetic acid (p-SCN-Bn-CHX-A″-DTPA) was procured from Macrocyclics (Dallas, TX), while immobilized pepsin resin used for enzymatic digestion was purchased from Thermo Scientific (Rockford, IL).
Other chemicals such as sodium bicarbonate, sodium acetate, arsenazo III, and Dulbecco's modified Eagle's medium (DMEM) were purchased from Sigma-Aldrich, while fetal bovine serum (FBS) was from GIBCO. The cell lines, SK-OV-3, SK-BR-3 (HER2 positive), and MDA-MB-231 (triple negative), were procured from the National Centre for Cell Science (Pune, India). PD-10 columns and AMICON Ultra centrifugal filter devices (molecular weight cut off [MWCO] 10,000 Da and MWCO 30,000 Da) were obtained from GE Healthcare and Millipore, respectively, for purification.
Radioactive counting was performed using a well-type NaI (Tl) scintillation counter (Electronics Corporation of India Limited). The size exclusion high performance liquid chromatography (HPLC) system (SE-HPLC; JASCO) equipped with a TSK gel column (G3000 SWXL) from Sigma-Aldrich was used for HPLC analyses. Ultraviolet-visible (UV/VIS) and radioactive detectors (Raytest GmbH), connected to the HPLC system, were used for measuring absorbance and radioactivity, respectively.
Analyses of radiochromatograms were performed using the GINASTAR software (Raytest GmbH). Ultraviolet (UV) absorbance measurements were carried out using a Chemito Spectrascan UV2600 spectrophotometer (Thermo Scientific). Guava flow cytometer kits were purchased from Merck KGaA (Darmstadt, Germany) and samples were acquired on the Guava flow cytometer.
Preparation and purification of pertuzumab F(ab′)2 fragments
Fragmentation of pertuzumab was carried out according to the manufacturer's protocol (Thermo Scientific). In brief, intact pertuzumab (∼20 mg/mL) was mixed with 1 mL of digestion buffer (sodium acetate, 20 mM, pH 4.5) and concentrated to 1 mL. The concentrated pertuzumab was added to 0.7 mL of pre-equilibrated immobilized pepsin (3000 U/mg). Postincubation, the reaction mixture was quenched by addition of 10 mM Tris-HCl buffer, pH 8.0, and centrifuged at 1000 g for 5 min.
The supernatant containing the F(ab′)2 fragments of pertuzumab was purified using the Amicon filtration device (MWCO 50,000 Da) and resuspended in 1 mL of phosphate buffer saline (PBS, 20 mM, pH 7.4). Pure F(ab′)2-pertuzumab was characterized by both SE-HPLC and gradient (5%–15%) sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE).
Conjugation of pertuzumab F(ab′)2 fragments with p-SCN-Bn-CHX-A″-DTPA
For radiolabeling with 177Lu, pertuzumab F(ab′)2 fragments were conjugated with p-SCN-Bn-CHX-A″-DTPA at a 1:10 molar ratio, as previously reported by the authors' group. 22 After the conjugation reaction, the conjugates were purified using the Amicon filtration device (MWCO 10,000 Da) to remove any unreacted CHX-A″-DTPA in the solution.
The concentration of F(ab′)2 fragments was determined by the Bradford method, 27 while the number of CHX-A″-DTPA molecules conjugated per pertuzumab F(ab′)2 fragment was determined by spectroscopic assay using Cu (II)-arsenazo (III). 28
Radiolabeling of CHX-A″-DTPA-F(ab′)2-pertuzumab conjugate with 177Lu
Radiolabeling of CHX-A″-DTPA-F(ab′)2-pertuzumab with 177Lu was performed as reported earlier, 22,29 wherein the immunoconjugates in 0.1 M sodium acetate solution (pH 5.6) were mixed with ∼185–370 MBq of 177LuCl3 (carrier added, specific activity ∼629–814 MBq/μg) and incubated at ambient temperature for 15 min. The reaction mixture was purified through a PD-10 column using 0.1 M sodium acetate solution (pH 6.0) as the eluent.
The radiochemical purity (RCP) of the radioimmunoconjugate was determined by SE-HPLC on a TSK gel column isocratically, using 0.05 M phosphate buffer, pH 6.8, for elution at a flow rate of 0.6 mL/min. RCP was concurrently ascertained by thin-layer chromatography (TLC) using 10 mM sodium citrate (pH 5.0) as the mobile phase.
Saturation binding assay of the radioimmunoconjugate
For the saturation binding assay, SK-OV-3, SK-BR-3, and MDA-MB-231 cells (1 × 106) were seeded in a six-well plate in triplicates. The required dilutions of the radioformulation were prepared in PBS with concentration ranging from 2.5 to 100 nM. After adding the required amount of antibody solution to each well, cells were incubated at 4°C for 2 h. Nonspecific binding was estimated by coincubating cells with 100-fold excess of unlabeled pertuzumab.
Subsequently, cells were washed thrice with ice-cold PBS and harvested using 0.5 N NaOH solution for counting the cell-associated activity in a well-type γ-counter. Data were analyzed by the least square regression method using GraphPad Prism 7.0 software, and Kd and Bmax values were evaluated for the radioformulation.
Determination of immunoreactive fraction
The total immunoreactive fraction (IRF) of the radiolabeled formulation was evaluated following a reported protocol by Lindmo et al., which is based on extrapolation of binding of the radiolabeled antibody at infinite antigen excess. 30 Briefly, SK-OV-3, SK-BR-3, and MDA-MB-231 cells (5 × 105–1 × 107) were harvested in 500 μL of DMEM (in triplicates). A fixed amount of radioformulation [∼20 nM of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab with ∼20,000 cps activity] was added to each tube containing cells and incubated at 4°C for 2 h with gentle shaking. Nonspecific binding was also assessed by addition of excess of cold antibodies.
Cells were washed twice with cold PBS, and radioactivity associated with cells was measured using the NaI (Tl) counter. The obtained data were then plotted as the reciprocal of the cell number (X-axis) against the reciprocal of the bound fraction (Y-axis). Furthermore, the data were then fitted according to a linear regression method using GraphPad Prism 7.0, and IRF was calculated using the equation {100 × (1/Y − intercept)} from the graph.
Studies to evaluate the binding synergy of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab and trastuzumab to the HER2 receptor
Competitive binding studies were performed with [177Lu]Lu-DTPA-F(ab′)2-pertuzumab in SK-OV-3, SK-BR-3, and MDA-MB-231 cells. Approximately 1 × 106 cells were seeded in six-well plates. The dilutions of immunoconjugate (1 × 10−12–6 × 10−6 M) were prepared in PBS, and cells were incubated with the required amount of unlabeled antibodies along with a fixed amount of radioimmunoconjugate for 2 h at 4°C. At the end of the incubation period, cells were washed thrice with ice-cold PBS and harvested using 0.5 N NaOH solution for counting the cell-associated activity in a well-type γ-counter.
Data were analyzed by a least square regression method and GraphPad Prism 7.0 software.
In vivo biodistribution studies
The animal studies were approved by the Institutional Animal Ethics Committee (IAEC) of BARC, and all animal experiments were carried out in strict compliance with the institutional guidelines, following the relevant national laws related to the conduct of animal experimentation. SK-OV-3, SK-BR-3, and MDA-MB-231 cells were injected (5 × 106 [5 million] cells in 100 μL) in SCID mice for inducing tumors. Tumor-bearing mice were assigned to two groups (n = 3), in which three animals from each group were injected with [177Lu]Lu-DTPA-F(ab′)2-pertuzumab (∼3.7 MBq/10 μg) in the presence/absence of excess of trastuzumab (∼20 × , ∼200 μg in 100 μL of saline).
The in vivo specificity and uptake of radiolabeled F(ab′)2-pertuzumab alone and in the presence of trastuzumab were determined in HER2-positive (SK-OV-3 and SK-BR-3) and -negative tumor (MDA-MB-231)-bearing mice at 24 and 48 h postinjection (n = 3).
Statistical analysis
An unpaired t-test was used for comparing the means of organ/tumor uptake studies in SCID mouse groups treated with [177Lu]Lu-DTPA-F(ab′)2-pertuzumab alone and in the presence of cold trastuzumab. One-way analysis of variance test was used to indicate the statistically significant difference between groups for % binding enhancement. The Tukey method was employed for comparing the mean of different groups.
Results
A detailed schematic view of the production, purification, and radiolabeling protocol of F(ab′)2-pertuzumab with 177Lu is depicted in Figure 1.

Schematic view of generation of radiolabeled F(ab′)2 fragments of pertuzumab.
Preparation and purification of pertuzumab F(ab′)2 fragments
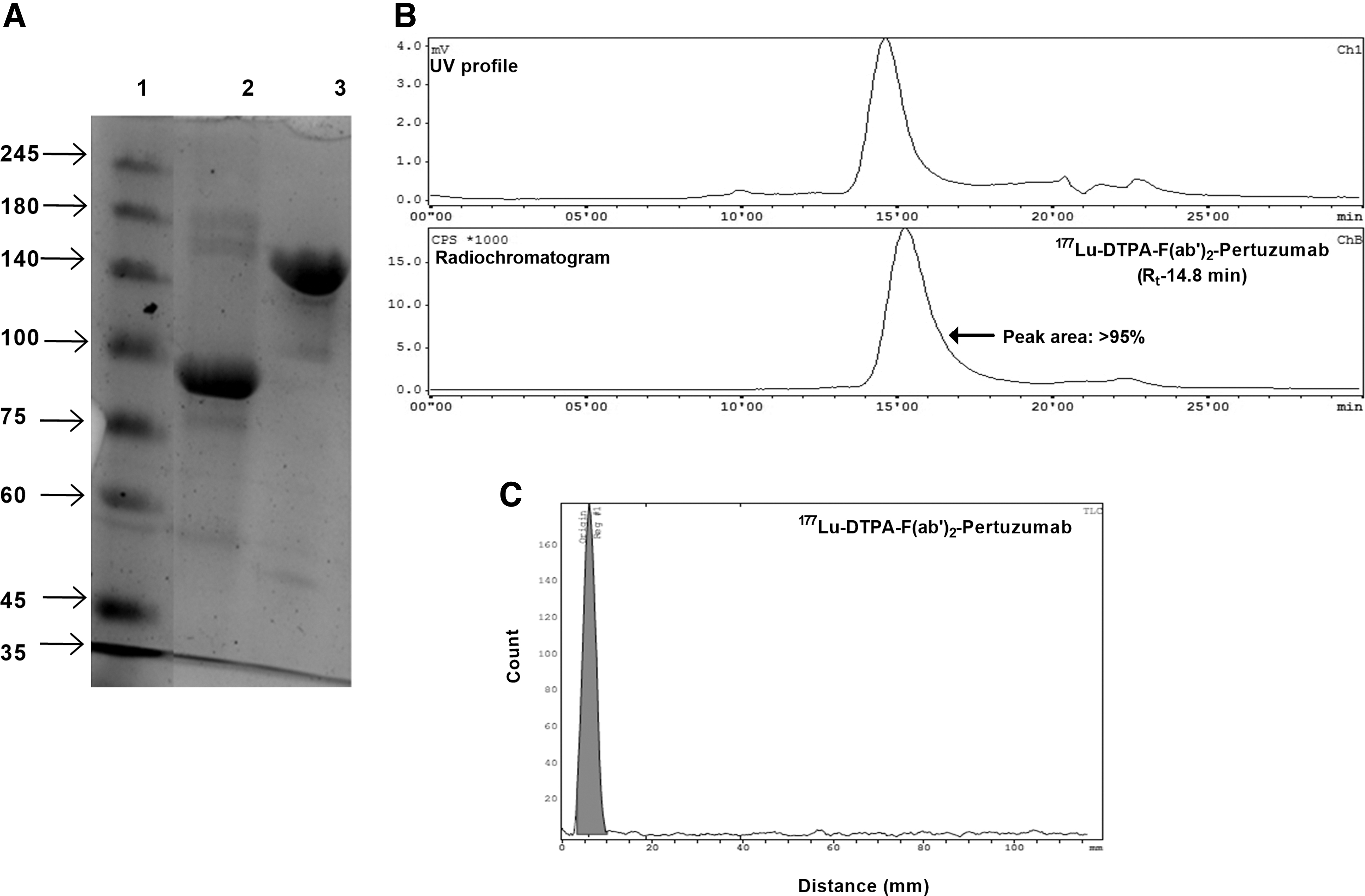
Pertuzumab F(ab′)2 fragments could be generated with maximum yield of 60%. The time required for complete digestion was found to be 18–24 h. The SDS-PAGE profile of intact and F(ab′)2 fragments of pertuzumab under nonreducing condition is shown in Figure 2A. A single band was observed corresponding to the molecular weight of ∼100 kDa (Fig. 2A, lane 2), confirming the purity of F(ab′)2-pertuzumab, whereas intact pertuzumab was resolved at ∼150 kDa in lane 3.
Conjugation of F(ab′)2-pertuzumab fragments with p-SCN-Bn-CHX-A″-DTPA
The average number of p-SCN-Bn-CHX-A″-DTPA molecules conjugated to F(ab′)2-pertuzumab was found to be 2.4 ± 0.35, as evaluated by the spectroscopic assay involving formation of the Cu-arsenazo (III) complex.
Radiolabeling of the CHX-A″-DTPA-F(ab′)2-pertuzumab conjugate with 177Lu
Radiolabeling was performed at ambient temperature with radiolabeling yield and RCP ranging from ∼70% to 90% before purification. The RCP of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab postpurification was determined by SE-HPLC. The complex was formed with >95% RCP postpurification with a retention time of ∼15.3 min (Fig. 2B, the radiochromatogram peak is >95%).
The SE-HPLC profile corroborated with TLC data wherein the entire radioactivity of the formulation was retained at the point of spotting ([177Lu]Lu-DTPA-F(ab′)2-pertuzumab) with negligible activity at the solvent front (free 177Lu) (Fig. 2C). The specific activity of radioformulations was found to be in the range of ∼50–125 MBq/nmoles. The radioformulation was found to be stable under different conditions up to 7 d postpreparation (Supplementary Table S1 and Supplementary Fig. S1).
IRF of 177Lu-DTPA-F(ab′)2-pertuzumab
The percentage immunoreactivity of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab was estimated using a Lindmo plot in HER2-overexpressing cell lines. The maximum reactive fraction was found to be 81.3% and 74.6% (Fig. 3A), respectively, in SK-OV-3 and SK-BR-3 cell lines, indicating robust receptor binding under in vitro conditions.

The reactive fraction in MDA-MB-231 was found to be 18.2%, indicating the nonspecific binding of the radioimmunoformulation. The reactive percentage with increasing cell concentration (5 × 105–1 × 107) varies from 11% to 68% for SK-OV-3, 11% to 73% for SK-BR-3, and 10% to 20% for MDA-MB-231 cells (Fig. 3B).
Saturation binding assay of radioimmunoconjugate
A strong binding affinity was revealed through a saturation binding assay performed in vitro in HER2-overexpressing cell lines. The dissociation constant (Kd) was found to be 18.19 ± 2.95 nM and 17.03 ± 2.99 nM in SK-OV-3 and SK-BR-3 cells, respectively (Fig. 3B). No specific binding (saturation) was observed in HER2-negative cells (MDA-MB-231).
Binding synergy of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab and trastuzumab to the HER2 receptor
The binding synergy was assessed through a competitive binding assay where uptake of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab was increased by 24.80% ± 7.45% and 22.96% ± 4.94% in SK-OV-3 and SK-BR-3 cells, respectively, when coincubated with excess of cold trastuzumab (Supplementary Fig. S2, p < 0.05, n = 3). Similar binding enhancement to 25.0% ± 7.07% and 26.00% ± 2.82% for SK-OV-3 and SK-BR-3 cells, respectively, was observed when cells were treated with excess of unlabeled F(ab′)2-trastuzumab (Supplementary Fig. S2, p < 0.05, n = 3).
No significant difference in binding enhancement was observed between SK-OV-3 and SK-BR-3 cells, indicating a similar receptor expression and binding synergy pattern in both cell lines (Supplementary Fig. S2, p > 0.05). In the presence of excess cold pertuzumab, competitive inhibition was observed in both cell lines (Fig. 4A–D).

Competitive binding assay with increasing concentration of unlabeled pertuzumab/trastuzumab and their F(ab′)2 fragments with [177Lu]Lu-CHX-A″-DTPA-F(ab′)2-pertuzumab in HER2-overexpressing
In vivo biodistribution studies
The biodistribution profile revealed significant tumor uptake and retention of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab in HER2-overexpressing tumor-bearing SCID mice. The predominant route of clearance was through kidneys while considerable uptake in the liver was also observed. The higher uptake of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab in the spleen is unique in SCID mice owing to the absence of an immunogenic response (e.g., the lack of T and B cell functions) and catabolic activity by macrophages in the spleen. 31 A high splenic uptake of the radiolabeled antibody was also observed in immunodeficient NOG mice. 32,33
Interestingly, in the authors' studies, despite lacking the Fc portion, the splenic uptake of F(ab′)2-pertuzumab was significantly higher. However, a higher spleen uptake has been reported for radiolabeled F(ab′)2 fragments by different groups. For instance, Wong et al. 34 reported an uptake of 5.43% ± 1.64% for [111In]In-CHX-A″-DTPA-F(ab′)2-panitumumab, while much higher splenic uptake of ∼20%–30% injected dose/g was observed with [89Zr] Zr-Df-F(ab′)2-anti-PD-L1 at different time points in wild-type C57bl/6 mice. 35 Further studies are desired to evaluate the pharmacokinetic behavior of humanized antibodies and their fragments in different preclinical animal models.
The maximum uptake of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab was observed at 24 h postinjection in each group, which decreased significantly at 48 h confirming its faster clearance from the circulation. The uptake was found to be higher when mice were preinjected with excess of unlabeled trastuzumab; however, there was no significant difference (p > 0.05, n = 3) compared with SK-OV-3 tumor-bearing mice injected with [177Lu]Lu-DTPA-F(ab′)2-pertuzumab, indicating no significant in vivo synergy in terms of tumor uptake at 24 h (Fig. 5A, B).

Ex vivo biodistribution and tumor uptake study with [177Lu]Lu-CHX-A″-DTPA- F(ab′)2-pertuzumab in SCID mice bearing SK-OV-3 and SK-BR-3 tumors at
Interestingly, at 48 h postinjection, tumor uptake of the radiotracer was significantly enhanced in mice pretreated with cold trastuzumab compared with untreated mice (p > 0.05, n = 3) (Table 1). In case of SK-BR-3 tumor-bearing mice, no significant tumor uptake enhancement was observed at 24 and 48 h (Fig. 5C, D, [p > 0.05, n = 3]). The specificity of tumor uptake of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab was also evaluated in HER2-negative (MDA-MB-231) tumor-bearing SCID mice wherein tumor uptake was found to be significantly less ([3.66 ± 3.28] %; Supplementary Fig. S3; p < 0.05, n = 3) at 48 h postinjection, while in the presence and absence of cold trastuzumab, no significant difference was observed in tumor uptake ([3.92 ± 2.99] %, [p > 0.05, n = 3]) in MDA-MB-231 tumor-bearing animals (Supplementary Table S2).
Ex Vivo Biodistribution Profile in SK-OV-3 Tumor-Bearing SCID Mice Injected with [177Lu]Lu-F(ab′)2 Fragments
SCID, severe combined immunodeficient.
Discussion
Radiolabeled monoclonal antibodies such as trastuzumab and pertuzumab hold promise as an effective therapeutic approach, particularly for HER2-positive cancers that have become resistant to immunotherapeutic agents. Numerous preclinical and clinical studies have reported encouraging results in targeting aggressive breast and ovarian cancer subtypes using these radiolabeled antibodies. 15 –19 However, these formulations suffer from certain limitations, such as slow pharmacokinetics and poor penetration in solid tumor vasculature, resulting in a lower target-to-nontarget ratio and unnecessary radiation exposure to vital organs. 26,31
To address these limitations, researchers are exploring various treatment modalities to improve the target-to-nontarget ratio for the diagnosis and therapy of HER2-positive cancers. One such approach gaining attention is the use of antibody fragments as theranostic agents due to their relatively faster pharmacokinetics and deeper penetration in solid tumors. Additionally, studies are investigating the impact of dual epitope targeting on the overall efficacy of radiolabeled antibody fragments. Coadministration of unlabeled trastuzumab has shown potential in enhancing the binding or uptake of radiolabeled pertuzumab in HER2-positive tumors. 20 –22,31
In the present study, the 177Lu-labeled F(ab′)2 fragment of pertuzumab was formulated and evaluated as a potential theranostic agent through in vitro and in vivo experiments. This radioformulation demonstrated a strong affinity and reactivity toward target receptors, and its uptake in cells and tumors was studied both in the presence and absence of unlabeled trastuzumab, confirming the role of dual epitope targeting of HER2 receptors.
A robust clinical synergy has been observed in the combinatorial therapy for breast cancer using trastuzumab, pertuzumab, and docetaxel 12,36 ; however, the role of radiolabeled trastuzumab and pertuzumab in targeting the HER2 receptor simultaneously has not been explored in detail. Few computational studies have revealed preliminary evidence where trastuzumab binding to HER2 could enhance the binding of pertuzumab to HER2 receptors. 20 Interestingly, Lua et al. reported no significant binding synergy when trastuzumab and pertuzumab were coincubated with pure HER2 receptors, indicating that HER2 receptors alone are not enough for such synergistic binding. 37
However, Marquez et al. demonstrated ∼30% enhancement in [89Zr]Zr-pertuzumab binding and tumor uptake in the presence of excess unlabeled trastuzumab, while Hao et al. reported a similar binding synergy with 67Cu-pertuzumab in HER2-overexpressing cell lines and tumor xenografts. 38 Recently, the authors' group conducted similar experiments with [177Lu]Lu-DTPA-pertuzumab in HER2-overexpressing cells and in tumor xenografts where binding synergy was observed under in vitro conditions and tumor uptake enhancement was revealed through single photon emission computed tomography imaging studies. 22
The fate of F(ab′)2 fragments and role of Fc regions of monoclonal antibodies under in vitro conditions were reported by Sharma et al. 21 where a significant binding synergy of [125I]I-F(ab′)2-pertuzumab in the presence of unlabeled trastuzumab was observed. In the present study, the enhanced cellular uptake of [177Lu]Lu-DTPA-F(ab′)2-pertuzumab was confirmed in HER2-positive cell lines in the presence of unlabeled trastuzumab, indicating the retention of binding synergy and dual epitope targeting in domains II and IV of the HER2 receptor.
Additionally, the role of the Fc region of the monoclonal antibody was found to be subtle under such interaction conditions since similar binding synergy was obtained with both [177Lu]Lu-DTPA-F(ab′)2-pertuzumab and [177Lu]Lu-DTPA-pertuzumab upon coincubation with F(ab′)2-trastuzumab or trastuzumab. 22 A comparative analysis of the two formulations is summarized in Table 2.
Ex Vivo Biodistribution Profile in SK-BR-3 Tumor-Bearing SCID Mice Injected with 177Lu-Labeled F(ab′)2 Fragments
SCID, severe combined immunodeficient.
Previously, Sharma et al. reported significant tumor uptake enhancement with intact pertuzumab radiolabeled with 177Lu. 22 In the present study, significant in vitro binding enhancement (∼20%–30%) was observed with HER2-overexpressing cells in the presence of trastuzumab/F(ab′)2-trastuzumab. Under in vivo conditions, the tumor uptake enhancement was not found to be significant in SK-BR-3 tumor-bearing mice.
However, the SK-OV-3 tumor model revealed a lower but significant tumor uptake enhancement at 48 h. This difference in in vitro and in vivo binding/tumor uptake could be attributed to the absence of physiological factors under in vitro conditions, which offer high binding probabilities compared with corresponding in vivo conditions.
Additionally, under in vivo conditions, immune and physiological clearance and the tumor microenvironment often limit the availability of radioformulations at the target site. [177Lu]Lu-DTPA-F(ab′)2-pertuzumab was cleared much faster from circulation compared with intact radiolabeled pertuzumab and this could be the potential explanation for the lack of significant tumor uptake enhancement with [177Lu]Lu-DTPA-F(ab′)2-pertuzumab in the presence of cold trastuzumab.
Tumor penetration of radiolabeled antibodies is hampered by physiological barriers, such as a binding site barrier and high pressure in the tumor interstitium, 39 –41 and these can be circumvented by the use of F(ab′)2 fragments for theranostic applications (Table 3). The [177Lu]Lu-DTPA-F(ab′)2-pertuzumab formulation was stable in biological fluids and exhibited high specificity and affinity toward HER2 receptors, thus the radioformulation could be employed as a theranostic radiopharmaceutical for HER2-overexpressing cancers.
Comparative Analysis of [177Lu]Lu-DTPA-Pertuzumab and [177Lu]Lu-DTPA-F(ab′)2-Pertuzumab
HER2, human epidermal growth factor receptor 2; ID, injected dose.
Additionally, there is rise in studies involving dual epitope receptor targeting (insulin-like growth factor-I receptor and death receptor) 42,43 to achieve maximum therapeutic efficacy and such strategies could be coupled with radiolabeled fragments to further enhance the pharmacokinetics, targeting, penetration, and retention of radioformulation within the tumor volume.
While the present experiments provide evidence of in vitro and, to some extent, in vivo binding synergy between [177Lu]Lu-DTPA-F(ab′)2-pertuzumab and trastuzumab, further studies are required to fully exploit the benefits of these strategies in cancer management. Investigating the structural and conformational details of binding synergies could enhance the concept of dual epitope targeting of HER2 receptors using radiolabeled antibody-based formulations.
Moreover, employing radiolabeled bispecific or multispecific antibodies that target two or more epitopes of the same or different receptors could be a crucial strategy for the management and treatment of solid tumors.
Conclusions
The radioformulation, [177Lu]Lu-DTPA-F(ab′)2-pertuzumab, offers a promising avenue for targeting HER2-positive cancers with radionuclide therapy. The utilization of dual epitope targeting by incorporating cold trastuzumab presents an attractive strategy to augment the therapeutic effectiveness of this approach. Further exploration and evaluation of this combined strategy could lead to significant advancements in the treatment of HER2-positive malignancies.
Footnotes
Acknowledgments
Authors are grateful to the staff of the Radiochemicals Section, Radiopharmaceuticals Division (RPhD), BARC, for supplying Lu-177 for the studies. Help rendered by Ms. Swati Dhavale in animal experimentation is thankfully acknowledged.
Authors' Contributions
R.S. was involved in conceptualization, radiolabeling, and characterization studies; A.M. was involved in conceptualization, animal experimentation, and manuscript preparation; and A.K. and H.D.S. contributed in animal experiments. All coauthors have reviewed and approved the manuscript before submission.
Disclosure Statement
No competing financial interests exist.
Funding Information
Research at the BARC is part of the ongoing activities of the Department of Atomic Energy, India, and is fully supported by government funding.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.