
Abstract
Background:
Bone metastasis (BM) is a serious clinical symptom of advanced colorectal cancer. However, there is a lack of effective biomarkers for early diagnosis and treatment.
Method:
RNA-seq data from public databases (GSE49355, GSE101607) were collected and normalized and batch effects were removed using the combat package. Differential expression analysis was performed to identify significant genes. Robust Rank Aggregation and machine learning algorithms were used to pinpoint candidate biomarkers. These biomarkers were validated using immunohistochemistry and further analyzed for survival rates. Enrichment analysis was conducted to explore biological mechanisms. Additionally, drug sensitivity and immune infiltration analyses were performed to provide insights into potential therapeutic targets.
Results:
Analysis results revealed 386 genes elevated in primary versus normal tissues and 26 genes varying between primary and BM. Serpin Protease Inhibitor Clade H1 (SERPINH1) as a novel biomarker for colon cancer metastasis. High SERPINH1 expression correlates with poor survival outcomes and is linked to high lymphatic invasion and advanced cancer stages. Additionally, SERPINH1 expression influences immune infiltration and is not predictive of chemotherapy response, but potential new drugs are suggested for high-expression cases. The gene also enriches classical cancer pathways such as Hedgehog and transforming growth factor-β.
Conclusions:
We identified novel colon cancer BM markers, including SERPINH1, using machine learning algorithms combined with traditional transcriptomic data and validated their expression through immunohistochemistry. This biomarker could significantly assist clinicians in making more precise treatment decisions.
Introduction
Colorectal cancer is one of the most common malignancies worldwide and its incidence has been on the rise in recent years. Bone metastasis (BM) is a severe clinical manifestation in the advanced stages of colorectal cancer. 1 Although it is relatively rare in clinical practice, when it occurs, patients often suffer from severe complications such as intense bone pain, significantly affecting their quality of life. Early diagnosis and treatment are crucial for improving the clinical prognosis of these patients. However, there is currently a lack of effective biomarkers for early diagnosis and treatment. 2 Therefore, in-depth research into the risk factors and prognostic factors for bone metastases caused by colorectal cancer is extremely important for enhancing understanding of such late-stage malignant events. This research could significantly assist clinicians in making more precise treatment decisions.
In recent years, machine learning technology has demonstrated significant innovation and potential in screening tumor markers compared to conventional histology techniques. 3,4 This technology uses advanced algorithms to extract a large number of complex oncological features from conventional transcriptome sequencing data and has been widely used in the diagnosis of many types of cancers, and the identification of potential targets. For example, Shi et al. developed a panel of three miRNA signatures to improve the diagnostic efficacy of pancreatic cancer using four machine-learning algorithms. In addition, the team also developed a new potential biomarker COL11A1 for breast cancer, which provides new insights for breast cancer treatment. 5,6 These discoveries have been made possible by the ability of machine learning to identify patterns of gene interactions and potential associations that are difficult to detect with traditional methods, thus providing more personalized decision support for cancer treatment. However, while machine learning has proven its effectiveness in other cancer types, its specific value and potential for application in colorectal cancer BM is still in the exploratory phase. As a complex clinical problem, colorectal cancer BM involves multifaceted interactions between tumor cells and bone tissues and precise and in-depth analyses are needed to reveal its possible biological mechanisms. 2
In this current study, our team used several machine learning algorithms that integrate transcriptomic data to identify potential biomarkers of BM in colon cancer and predicted potential Food and Drug Administration (FDA)-approved drugs based on these markers, providing new insights to enable precision therapy in this population.
Materials and Methods
Data collection and prepared
We collected RNAseq data from the GEO database, the issue numbers are GSE49355 and GSE101607 as training datasets. The GSE49355 database includes 18 normal colon tissues, 20 primary colon cancer tissues, and 19 colon cancer liver metastasis tissues. In GSE49355, we only selected two colon cancer BM samples. The data were normalized by the limma package and the batch effect was removed by the combat package. The validation data from the public database and our hospital colon cancer center. The radiomics-related CT data were normalized by Python (Version 3.8).
Remove batch effect and conduct different expression analysis
We used R package combat to remove the batch effect between two datasets, GSE49355 and GSE101607 (two colon cancer BM samples). Then, we conducted the differential expression genes (DEGs) analysis between normal and primary colon cancer tissues, primary colon cancer and liver metastasis tissues, and liver metastasis tissues versus BM tissues. The absolute value of logFC more than 1 with a p-value < 0.05 was identified as a significant gene.
Robust rank aggregation and Machine learning method identify candidate biomarker
We used robust rank aggregation (RRA) algorithms to identify the important gene between three different expression genes results. To ensure the result is more credible, we also performed the random forest (RF) and support vector machine (SVM) algorithms to select the feature DEGs genes and resource the BM and no BM tissues. We used parameter optimization to determine the optimal parameter values for the machine learning model and the random seed was set to 2024. Afterward, we merge them with the Venn plot to select the candidate biomarkers, which are associated with colon cancer BM.
Validate the expression of the candidate and analyze survival difference
We used the public database The Cancer Genome Atlas Program Colon adenocarcinoma (TCGA–COAD) to validate the expression of hub gene in transcriptional levels to protein level and also to validate expression in different colon cancer cell lines. The endpoint was overall survival (OS), disease free syrvival (DFS), and progress free interval (PFI) and the survival analysis was conducted by log-rank test, the best cutoff value of expression was identified by the survminer package.
Candidate biomarker with clinical factor
We also analyzed the candidate biomarker expression in patients’ baseline information (gender, race, and body–mass index [BMI]) and another important clinical factor (N stage, T stage, AJCC stage, lymphatic invasion, carcinoembryonic antigen (CEA) level, residual tumor, and primary therapy outcome), which are also associated with the patient’s outcome. The different analyses between groups were conducted by the Wilcoxon test.
Immune infiltration analysis of candidate biomarker
We used the CIBERSORT package to perform immune infiltration analysis, which involves calculating the stroma score, immune score, and estimate score. Additionally, we examined the expression relationship in different immune cells and conduct correlations between this marker and normal immune checkpoints (CD274, PDCD1, and CD86). Furthermore, we investigated the relationship between this marker and MSI, Homologous Recombination Deficiency (HRD), and neoantigen.
Durg sensitive analysis of this biomarker
We collected the normal chemotherapy drugs name list from the Genomics of Drug Sensitivity in Cancer (GDSC) database, then, we used the R package prophetic to conduct the Durg sensitive analysis, the p-value < 0.05 were identified as significant. In addition, to provide more therapy plans for clinical decisions, we also use this database to predict some new FDA-approved candidate drugs for this target.
Biomarker with bone metastasis-related radiomics features
We collected the colon cancer patients with BM and used 3D slice to extract related radiomics features (Step 1. Marked the tumor side in each images to obtain the mask files; Step 2. Used python to extract radiomics features.) Then, we conducted machine learning algorithms, principal component analysis (PCA), and Lasso regression to select the important features, after merge both algorithms results to identify the common BM-related features. Finally, we performed the correction analysis between biomarker and above features.
Gene oncology and pathway enrichment analysis
We used ClusterProfile to conduct the enrichment analysis with Gene Oncology to explore the candidate Biological mechanisms. It included Biological Processes, Molecular Functions, and Cellular Components. We also performed the pathway enrichment to understand the regulatory mechanisms of these genes in organisms.
Results
Significant differential expression gene between colon cancer and metastasis tissues
Before analysis, we removed the batch effect from two different datasets and after conducting, we found that the data had reached normalized level, while comparing the previous datasets at an independent level (Fig. 1A, B). Then we conducted the DEGs analysis, we found that 386 genes had high expression in primary colon cancer tissues when compared with normal colon tissues (Fig. 1C). In addition, when comparing primary colon cancer tissues with colon cancer liver metastasis tissues, 146 gene expression is high in colon cancer liver metastasis groups (Fig. 1D). Moreover, we also found that a total of 26 genes, including 14 upregulation genes and 12 downregulation genes, are different expressions between primary colon cancer tissues and BM tissues (Fig. 1E).

Identification of SERPINH, a gene associated with BM in colon cancer. Before and after removal of batch effect in different datasets
Serpin protease inhibitor clade H1 is a novel biomarker for colon cancer metastasis
We used RRA algorithms to identify the important and robust DEGs from the above results and the top 15 genes are shown in Figure 1F. To identify the biomarker for colon cancer metastasis, we also used RF and SVM to perform the feature selection between primary colon cancer tissues and BM tissues. The top 10 RF results are shown in Figure 1G and the SVM demonstrates that 17 features are identified (Fig. 1H), afterward, we merged RRA, RF, and SVM results and we found that only Serpin protease inhibitor clade H1 (SERPINH1) is a shared gene. So, we finally confirm that SERPINH1 is a novel biomarker associated with colon cancer metastasis (Fig. 1I).
Serpin protease inhibitor clade H1 is high expression in colon cancer tissues and cell lines
We used the extended database to validate the expression of SERPINH1 in colon cancer tissue and cell lines. The results show that no matter whether paired or not, SERPINH1 has high expression in colon cancer tissues when compared with normal tissues (Fig. 2A and B). At the protein level, we also demonstrated this conclusion (Fig. 2C and D). In addition, we also found that SERPINH was highly expressed in colon cancer cell line C2BBE1 (Fig. 2E).

Expression validation and survival analysis of SERPINH. At the transcriptome level, SERPINH was highly expressed in colon cancer tissues (
High expression serpin protease inhibitor clade H1 is associated with poor outcome of colon cancer
To evaluate the survival difference in three endpoints OS, DSS, and PFS, we used a log-rank test to perform this procedure. The survival analysis shows that compared with low-expression patients, high-expression SERPINH1 patients always with a poor outcome (Fig. 2F–H).
Serpin protease inhibitor clade H1 is associated with clinical factor
The baseline information and oncology factor will affect the patient’s outcome. We analyzed the SERPINH1 expression with this clinical factor and we found that in the baseline information group, the SERPINH1 expression was without difference between gender, race, BMI, and colon polyps present (Fig. 3A–C). However, in the oncology factor group, this gene expression difference in N stage, T stage, AJCC stage, and perineural invasion. In addition, we also demonstrated that high expression of SERPINH1 means high lymphatic invasion and tumor status after surgery (Fig. 3E–J). This gene is not associated with CEA level and though exploring the disruption of SERPINH and therapy response status, we also found this gene could not predict drug response in clinical application (Fig. 3K and L).
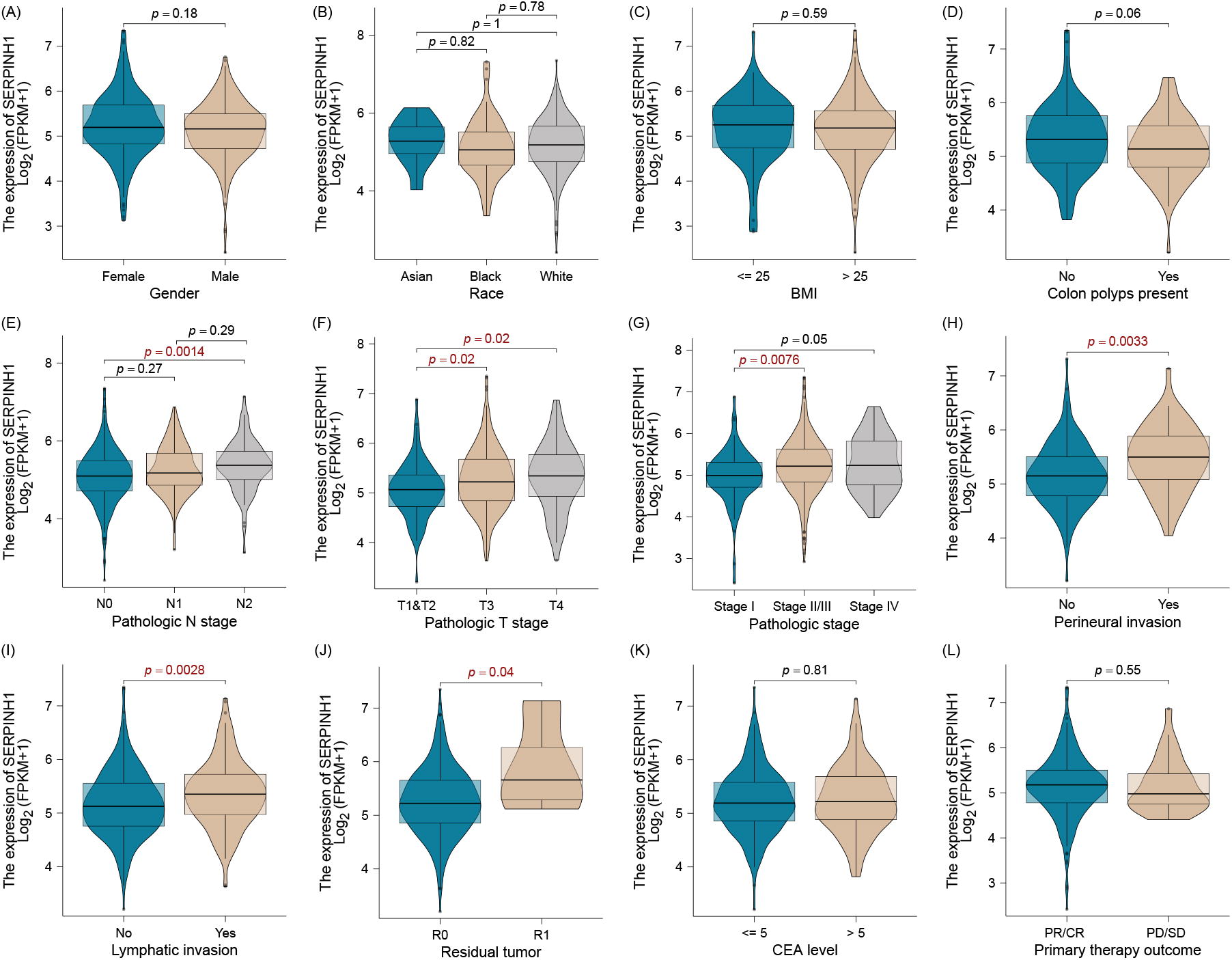
Clinical correlation between SERPINH, a gene related to BM in colon cancer. The SERPINH1 expression was without difference between gender, race, BMI, and colon polyps present
High expression serpin protease inhibitor clade H1 is associated with immune infiltration
The Cibersort algorithm’s results show that high expression of SERPINH1 is positively associated with stromal score, immune score, and estimated score (Fig. 4A–C). We also found that high expression of this gene is positively associated with CD4 native cells, macrophages M2, and eosinophils (Fig. 4D). Additionally, we found that high expression of SERPINH1 is positively associated with immune checkpoints and homologous recombination deficienc (HDR) (Fig. 4E–H), but not with Microsatellite instability (MSI) and neoantigen (Fig. 4I, J).

Colon cancer BM-related gene SERPINH and tumor immune infiltration. The high expression of SERPINH1 is positively associated with stromal score, immune score, and estimated score
Low expression serpin protease inhibitor clade H1 is not sensitive to the normal chemotherapy drug
Metastatic colon cancer also recommends the normal chemotherapy plan. Here, we evaluated the normal chemotherapy drug sensitivity in high- and low-expression SERPINH1 patients. We found that the patients with low SERPINH1 expression are not sensitive to Gefitinib, Cisplatin, Docetaxel, Axitinib, and Lapatinib (Fig. 5A–E). For future exploratory analysis, we recommend PD-0332991, BMS-708163, GW-441756, and OSI-906 as new FDA-approved candidate drugs for these patients (Fig. 5F–1).
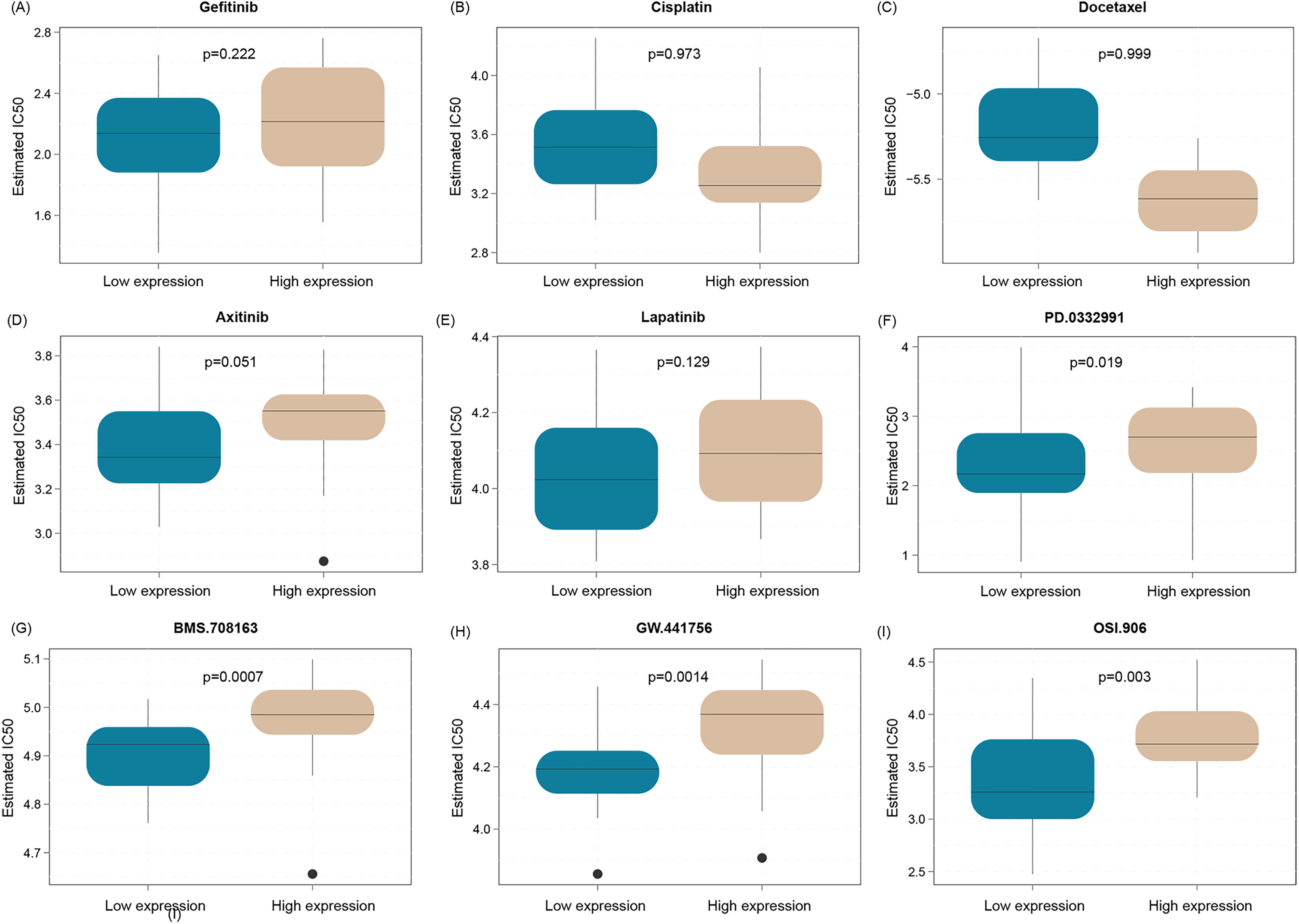
Predicted chemosensitivity of SERPINH, a gene associated with BM in colon cancer. The patients with low SERPINH1 expression are not sensitive to Gefitinib, Cisplatin, Docetaxel, Axitinib, and Lapatinib
Serpin protease inhibitor clade H1 is associated with bone metastasis-related radiomics features
The demo figure for this part analysis is shown in Figure 6A, the CT imaging of primary colon cancer and BM (Fig. 6B). Our study involved the collection of 25 paired primary colon cancer CT scans and BM CT scans, with the baseline information of enrolled patients depicted in Figure 6C. Following the application of PCA and Lasso regression models for feature extraction and selection, we identified 27 and 18 BM-related CT features, respectively. The subsequent merging of results from both algorithms revealed the presence of five common features (Fig. 6D and E). The correlation analysis between SERPINH1 and five BM-related features indicates that SERPINH1 exhibits a positive association with four of the BM-related features and a negative association with one of them (Fig. 6F).

The correlation of SERPINH1 and BM-related radiomics features. The demo figure for this part analysis
High expression serpin protease inhibitor clade H1 could enrich in classical signal pathway
The results of SERPINH1 single gene GSEA enrichment analysis suggested that high expression SERPINH1 could be significantly enriched into a variety of classical cancer-associated signaling pathways, such as Hedgehog signaling pathway and transforming growth factor-β signaling pathway (Supplementary Fig. S1A–F).
Discussion
Among individuals diagnosed with colorectal cancer, 20% are found to have metastatic colorectal cancer (mCRC) and 40% experience recurrence after initial treatment. 7,8 CRC typically spreads to the liver, lungs, and peritoneal cavity, making these metastases the main causes of CRC-related deaths. However, BM is rare, occurring in only 6%–10% of cases 9,10 and has often been overlooked due to its rarity and limited research data. Recent years have seen an increase in reported BM from CRC, likely due to extended survival times from new treatment regimens and advancements in diagnostic imaging techniques. To our knowledge, this study is the first to employ machine learning techniques to identify a hub gene (SERPINH1) predictive of BM occurrence and prognosis in CRC patients, validated through radiomics. This approach holds significant potential for the early diagnosis and treatment of BM in CRC patients.
The SERPINH1 gene belongs to the serpin superfamily, known for its protease inhibitory functions. SERPINH1 encodes the Heat Shock Protein 47 (HSP47), a molecular chaperone essential for the proper folding and secretion of collagen. This protein ensures the stability and correct assembly of collagen molecules within the endoplasmic reticulum. Aberrant expression of SERPINH1 has been detected in various cancers, such as lung adenocarcinoma (LUAD), 11 clear cell renal cell carcinoma (ccRCC), 12 breast cancer, 13 and CRC metastasis. 14 In a previous study, SERPINH1 was found significantly overexpressed in LUAD tissues and correlated with poor patient prognosis using data from the TCGA and a single-center LUAD cohort. Analysis also revealed that high SERPINH1 expression is linked to increased tumor mutation burden, distinct immune infiltration characteristics, and improved responses to immunotherapy and antitumor treatments, suggesting novel clinical management strategies for LUAD. 11 In another study, SERPINH1 was also identified as a significant biomarker for poor prognosis in ccRCC through integrated proteomic and transcriptomic screening, showing its strong association with transforming growth factor-β levels and epithelial-to-mesenchymal transition. Validation in two independent cohorts confirmed elevated SERPINH1 levels in ccRCC tissues and its role as an independent predictor of overall and disease-free survival, particularly in von Hippel–Lindau wild-type patients. 12 A study on CRC lymph node metastasis identified HSP47 as a novel biomarker, with higher expression in CRC tissues and an increased number of HSP47-positive spindle cells in the tumor stroma correlating with tumor progression. These spindle cells independently predict lymph node metastasis, early recurrence, and poor prognosis in CRC patients. 14 Our study results demonstrate that SERPINH1 is highly expressed in both the transcriptome and proteome of CRC tumor tissues and is associated with poorer patient prognosis, aligning with previous research findings. 11 –13 However, in contrast to the findings by Mori et al., 14 which indicated no significant correlation between HSP47 protein expression and clinicopathological features in CRC patients apart from lymph node metastasis, our study demonstrates significant associations between SERPINH1 expression and several clinical and pathological features in CRC patients, such as pathological N stage, pathological T stage, overall pathological stage, perineural invasion, lymphatic invasion, and residual tumor status. The observed differences may stem from the fact that our study focused on transcriptomic data. Additionally, the use of different cohorts in our research might have led to variations in expression profiles and their correlations with clinicopathological features in CRC patients. These methodological and cohort differences underscore the importance of considering both gene and protein expression levels, as well as the specific datasets used, in biomarker research.
Radiogenomics in CRC harnesses radiomics data to enhance genomic analysis, potentially serving as a “virtual biopsy” that can identify specific tumor regions with high mutation probabilities, thereby overcoming the limitations of traditional biopsy methods due to intratumoral heterogeneity. 15 This approach not only informs on genetic susceptibilities, such as resistance to anti-EGFR treatments, but also expands to predict therapy responses, prognostic outcomes, and metastatic potentials, primarily using CT-based models in clinical settings. 16,17 Recently, some scholars successfully developed a multiscale nomogram that integrates radiomics, genomic data, and clinical markers such as carbohydrate antigen 19–9 to predict metastasis in CRC patients, demonstrating high accuracy across different cohorts. The model shows significant potential for enhancing preoperative assessments and personalizing treatment strategies by providing reliable predictions of metastatic risk. 18 In another multi-institutional retrospective study, which included 1601 patients with CRC, scholars developed a radiogenomic prognostic model that links radiomic features with genomic subclones, substantially enhancing the prediction of DFS outcomes. By identifying and validating radiogenomic signatures associated with key biological pathways, the model serves as a robust tool that complements existing prognostic methods and significantly improves patient stratification based on survival probabilities. 19 As shown in our results, the significant correlation between SERPINH1 expression and the radiomic features we extracted for BM in CRC indicates its potential role. Additionally, the consistent expression of SERPINH1 across multiple CRC cell lines further supports its relevance and establishes a strong foundation for further research into SERPINH1 as both a predictive biomarker and a therapeutic target in CRC.
mCRC typically has a poor prognosis due to its advanced stage at diagnosis and the complexity of treatment options. Advances in molecular profiling have significantly enhanced the treatment of this condition by enabling therapies to be tailored to the specific genetic and biological characteristics of the tumor, thereby improving overall survival rates. 20,21 Particularly, targeted therapies and immunotherapy have shown promise in extending survival for patients with specific genetic mutations and microsatellite instability, respectively, although cures remain rare. For example, EGFR inhibitors, such as cetuximab and panitumumab, are effective exclusively for patients with KRAS/NRAS wild-type mCRC. 22 In the KEYNOTE-177 clinical trial, mCRC patients with MSI-H/Mismatch Repair Deficient could benefit from PD-1 blockade (Pembrolizumab) with longer PFS thanks traditional chemotherapy. 22 Interestingly, our study reveals that high expression of SERPINH1 is associated with a complex tumor microenvironment, increased immune cell infiltration, and elevated expression of immune checkpoint genes (CD274, PDCD1, and CD86). These factors collectively suggest that SERPINH1 may contribute to poor prognosis in CRC by promoting tumor–immune interactions that facilitate tumor progression and immune evasion. 23,24 Additionally, high association of SERPINH1 expression with HRD, 25 MSI, 26 and neoantigen presence, 27 further imply that SERPINH1 might influence the response to immunotherapy, highlighting its potential as a biomarker for predicting treatment outcomes and the combined use of SERPINH1-targeted inhibitor and checkpoint inhibitors may potentially benefit patients with mCRC. However, this should be validated in more preclinical studies.
Our study still has some limitations. First, our radiomics dataset was limited and we did not validate our model using radiogenomic results in a larger cohort. Second, although we found that SERPINH1 may be involved in immune evasion by tumor cells, the tumor microenvironment is complex and we did not explore its relevance at the single-cell level. Finally, more preclinical studies are needed to investigate the mechanisms of SERPINH1 in CRC BM and to determine whether it can serve as a therapeutic target.
In summary, our investigation identified the gene SERPINH1 is associated with BM in CRC patients and its elevated expression in patients with BM was further validated through radiomics analysis. Notably, SERPINH1 expression positively correlates with multiple immune checkpoints, suggesting its potential role in mediating immune evasion and promoting tumor metastasis. Thus, combining immunotherapy with SERPINH1 inhibitors may offer therapeutic benefits for this group of patients.
Footnotes
Authors’ Contributions
The conceptualization of the study was led by J.Z. and H.Z., while the methodology was developed by G.Z., T.S., and Q.Q. Q.Q. also conducted the formal analysis. G.Z. was responsible for preparing the data. J.Z. drafted the original article and, along with H.Z., supervised the entire project. The article revisions were undertaken by J.Z., who also managed the project administration and funding acquisition alongside H.Z. All authors have reviewed and approved the final version of the article.
Data Availability
All data can be obtained from the corresponding author.
Ethics Approval Statement
Ethics approval by the Guilin People’s Hospital.
Patient Consent Statement
Patient sign permission form and consent the data obtain.
Permission to Reproduce Material from Other Sources
All code with analysis can be obtained from the corresponding author.
Disclosure Statement
All authors declare no conflicts of interest.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.