
Abstract
Abstract
Based on microarray data comparing gene expression of fibroblast donor cells and bovine somatic cell nuclear transfer (SCNT) and in vivo produced (AI) blastocysts, a group of genes including several transcription factors was selected for evaluation of transcript abundance. Using SYBR green-based real-time polymerase chain reaction (Q-PCR) the levels of POU domain class 5 transcription factor (Oct4), snail homolog 2 (Snai2), annexin A1 (Anxa1), thrombospondin (Thbs), tumor-associated calcium signal transducer 1 (Tacstd1), and transcription factor AP2 gamma (Tfap2c) were evaluated in bovine fibroblasts, oocytes, embryos 30 min postfusion (SCNT), 12 h postfertilization/activation, as well as two-cell, four-cell, eight-cell, morula, and blastocyst-stage in vitro fertilized (IVF) and SCNT embryos. For every gene except Oct4, levels of transcript were indistinguishable between IVF and SCNT embryos at the blastocyst stage; however, in many cases levels of these genes during stages prior to blastocyst differed significantly. Altered levels of gene transcripts early in development likely have developmental consequences downstream. These results indicate that experiments evaluating gene expression differences between control and SCNT blastocysts may underestimate the degree of difference between clones and controls, and further offer insights into the dynamics of transcript regulation following SCNT.
Introduction
Incomplete nuclear reprogramming following SCNT is manifest in a variety of ways. Low rates of development and pregnancy establishment (Hill et al., 2000; Powell et al., 2004) as well as high rates of pregnancy failure throughout gestation (Heyman et al., 2002) and frequent postnatal loss are obvious indicators of deficiency in the SCNT process. Additionally, a variety of abnormalities can be observed in SCNT embryos, fetuses, placentas, and neonates. Differences in cell number and cell allocations have been observed in SCNT embryos as well as increased incidence of aneuploidy and fragmented nuclei (Booth et al., 2003; Li et al., 2004a; Pfister-Genskow et al., 2005). Following embryo transfer and pregnancy establishment a number of factors likely contribute to the high rates of pregnancy failure. Abnormal placentation has been reported in a number of species following SCNT (Constant et al., 2006; Fletcher et al., 2007; Hashizume et al., 2002; Ogura et al., 2002). In addition, fetal overgrowth (Constant, et al., 2006), hydroallantois (Lawrence et al., 2005), stillbirth, respiratory and circulatory problems (Hill et al., 1999), and liver malformations have all been observed in clones (Li et al., 2005). Many of these problems may be the direct result of the primary problem of improper placentation.
A number of studies have undertaken to characterize abnormal gene expression patterns following SCNT; however, the majority of the studies have evaluated gene expression in blastocysts, fetal tissues, or placental tissues. Many studies have evaluated the expression levels of specific genes important in early development in SCNT blastocysts by quantitative-polymerase chain reaction (Q-PCR). The list of genes reported to be differentially expressed in SCNT blastocysts includes Mash2, Dnmt1, Hsp 70.1, Ifnτ, Cx43 (Niemann et al., 2002), Oct4 (Boiani et al., 2002), G6pd, Xist, Pgk (Wrenzycki et al., 2002), several imprinted genes including Igf2 (Han et al., 2003), H19, and Snrpn (Mann et al., 2003), and many others. It is important to note that factors such as activation protocol, stage of donor cells, and culture conditions all impact gene expression in SCNT blastocysts (Wrenzycki et al., 2001).
Evaluation of global gene expression patterns in bovine SCNT blastocysts (Aston et al., 2009a; Beyhan, et al., 2007; Smith, et al., 2005; Zhou, et al., 2008), placental tissue (Everts et al., 2008; Oishi et al., 2006), and liver (Herath et al., 2006; Schrader et al., 2003) have also reported a number of differentially expressed genes. Surprisingly, these studies generally find fewer than 100 differentially expressed genes when compared with controls. In global gene expression studies little to no consensus exists in the lists of differentially expressed genes, likely a consequence of different SCNT protocols utilized by different researchers.
A recent report of global transcript abundance in mouse SCNT embryos during the first two cell cycles illustrates the importance of evaluating transcript abundance differences in early preimplantation embryos prior to the blastocyst stage to appreciate the scope of the problems in gene expression following SCNT. It was found that during the second cell cycle over 1000 genes were differentially represented between SCNT and control embryos, indicating the reprogramming process occurs over several cell cycles, and the divergence in gene expression patterns narrows greatly by the blastocyst stage (Vassena et al., 2007).
Although various studies report differential gene expression between SCNT and control tissues, they do not attempt to determine the point in development when expression levels in the SCNT tissues diverged from controls, nor do they address the question of timing of reprogramming events. These questions are important in elucidating mechanisms involved in epigenetic reprogramming following SCNT with the ultimate goal of improving the efficiency of SCNT. Two recent studies evaluated the mechanisms and timing of nuclear reprogramming globally following normal fertilization (Sun et al., 2007) and SCNT (Gao et al., 2007). Localization and activity of 20 different chromatin factors including transcription factors and transcriptional regulators was evaluated through early preimplantation development, and it was determined that in the case of normal fertilization and SCNT an “erase-and-rebuild” strategy for epigenetic modifications was employed. This strategy involves the global removal of chromatin factors prior to pronuclear formation followed by reassociation of the factors after pronuclear formation. Although the mechanisms of epigenetic reprogramming were found to be similar for IVF and SCNT embryos, the erasure of epigenetic marks as well as the reestablishment of new modifications was found to be both incomplete and delayed in SCNT embryos (Gao et al., 2007).
The inefficiencies associated with SCNT, numerous reports of abnormal epigenetic reprogramming manifest by gene-specific and global gene expression differences in SCNT embryos and fetal tissues, as well as the extremely limited understanding of epigenetic reprogramming mechanisms following SCNT all provided impetus for the present study. The aim of this study was to evaluate the dynamics of early embryonic transcript regulation by measuring the relative levels of transcript abundance through various stages of preimplantation development of several developmentally important genes known to undergo a high degree of change in expression following SCNT. This work was undertaken in an effort to gain insight into the timing of gene expression regulation following SCNT with the ultimate goal of elucidating reprogramming mechanisms. This is the first study to report detailed stage-by-stage transcript abundance in preimplantation bovine SCNT embryos.
Materials and Methods
Donor cell culture
Primary bovine fibroblast cultures were established from either lung tissue or ear biopsy. Previous data have demonstrated no difference in in vitro development between lung- and ear-derived donor cells (Kato et al., 2000). Tissues were washed thoroughly and minced, suspended in DMEM/Hams F12 (1:1) (Hyclone Laboratories, Logan, UT) supplemented with 15% fetal bovine serum (FBS; HyClone Laboratories) and 100 U/mL penicillin/100 μg/mL streptomycin (HyClone Laboratories), seeded in 25 cm2 tissue culture flasks, and cultured at 39°C in a humidified atmosphere of 5% CO2 in air for several days. Cells between passages one and four were then harvested and resuspended in tissue culture medium containing 10% DMSO, frozen, and stored in liquid N2 until use in microarray experiments or SCNT. Prior to gene expression studies cells were thawed and expanded from about three million cells to approximately 27 million cells through two passages. Cells utilized for SCNT were grown to 80–100% confluence then treated with trypsin (0.25%) and resuspended in manipulation medium prior to use.
Oocyte maturation
Maturation of bovine oocytes was performed as described previously (Li et al., 2004a, 2004b). Briefly, cumulus–oocyte complexes (COCs) were aspirated from 3–8 mm follicles using an 18-gauge needle from ovaries collected from a local abattoir. Only those oocytes with uniform cytoplasm and intact layers of cumulus cells were selected and matured in TCM 199 containing 10% FBS, 0.5 μg/mL FSH (Sioux Biochemicals, Sioux City, IA), 5 μg/mL LH (Sioux Biochemicals), and 100 U/mL penicillin/100 μg/mL streptomycin for 18–22 h.
SCNT embryo production
Following maturation, cumulus cells were removed from oocytes by vortexing COCs in PB1 [calcium- and magnesium-containing phosphate buffered saline (HyClone Laboratories), 0.32 mM sodium pyruvate, 5.55 mM glucose, 3 mg/mL BSA) medium containing 10 mg/mL hyaluronidase. Oocytes with a first polar body were used as recipient cytoplasts. Enucleation was employed to remove the first polar body and metaphase plate, and single cells were subsequently transferred to the perivitelline space of recipient cytoplasts. Fusions of NT couplets were performed in mannitol fusion medium (Wells et al., 1999) by two electric DC pulses of 2.2 kV/cm for 25 μsec. Following fusion, embryos were held in CR2 medium supplemented with 3% FBS for 1–2 h prior to activation (Rosenkrans and First, 1994). Fused embryos were activated between 23 and 25 h after the onset of maturation by exposure to 5 μM ionomycin for 5 min followed by 5 h incubation in 10 μg/mL cycloheximide. For the purposes of the microarray experiments we produced three groups of 10 grade 1–2 blastocysts from a single cell line. For the Q-PCR studies, three groups of five embryos each were collected at each embryonic stage to be analyzed. Embryos were placed in RNAlater RNA stabilization reagent (Ambion Inc., Austin, TX) and stored at −20°C until RNA extraction.
Artificial insemination (AI) embryo production
Control embryos for microarray studies were collected from superovulated cows using established protocols. Donor cows were synchronized using a vaginal progesterone implant. The implant was used for 10 days followed by an i.m. injection of 50 mg PGF2α (5 mL at 10 mg/mL). Animals were bred by AI the morning following standing heat and again 12 and 24 h after standing heat. Seven days after the initial breeding, embryos were collected from donor animals by intrauterine flush using embryo filters. Following collection embryos were rinsed in flush medium, placed in RNAlater, and stored at −20°C until RNA extraction. Three groups of 10 grade 1 and 2 blastocysts were collected for the microarray studies.
IVF embryo production
IVF embryos were collected for the Q-PCR component of the study. Cyropreserved bovine semen (Hoffman AI, Logan, UT) was thawed and live sperm were separated by centrifugation on a 45%/95% layered Percoll gradient. Motile spermatozoa obtained by this method were diluted in fert-TALP to a final concentration of 106/mL (Reed et al., 1996). Capacitation occurred in fert-TALP containing heparin at a concentration of 10 μg/mL. In vitro matured oocytes were fertilized in vitro for 18–20 h at 39°C in 5% CO2 and air. After the fertilization period, oocytes were vortexed in a 15-mL conical centrifuge tube containing 1 mL of PB1 2 min 40 sec to completely remove cumulus cells. Embryos were cocultured with cumulus cells in CR2 medium supplemented with 3% FBS (Rosenkrans and First, 1994) at 39°C in 5% CO2 in air. Three groups of five embryos each were collected at each embryonic stage to be analyzed. Embryos were rinsed through several drops of PB1 and through a single drop of RNAlater then placed in RNAlater and stored at −20°C until RNA extraction.
RNA extraction
RNA extraction from donor cells
Cells were harvested by trypsinization, washed with cell culture medium (DME/F12 1:1 supplemented with 15% Defined FBS and Penicillin/Streptomycin), followed by a second wash with PBS. Washed cells were pelleted and resuspended in RLT Buffer (Qiagen Inc., Valencia, CA) containing beta-mercapto ethanol (βME) and subsequently homogenized using a syringe with a 21-gauge needle. RNA extraction was performed using the RNeasy Mini RNA Extraction Kit (Qiagen) according to manufacturer's recommendations.
RNA extraction from embryos
Total RNA was extracted and DNA was digested with DNase I from AI, IVF, and NT embryos using the RNAqueous microkit
Microarray expression studies
For the embryo microarray studies previous experience, as well as personal communications with other researchers, indicated RNA concentration- and quality-determination using the nanodrop and bioanalyzer are ineffective with RNA extracted from embryos, so preliminary checks of RNA were not performed on embryonic RNA. Blastocyst stage bovine embryos contain approximately 2 ng total RNA, so to attain sufficient quantities of RNA for hybridization on Affymetrix GeneChips a two-round labeling protocol was used. After the two-round labeling procedure RNA quantity and integrity were assessed using an Agilent 2100 Bioanalyzer. Following quality assessment labeled RNA was hybridized to the Affymetrix bovine microarray chip and subsequently scanned according to manufacturer's protocols as described previously (Aston et al., 2009a). Microarray analysis of donor cells was also performed according to manufacturer's protocols. Because sufficient RNA could be obtained from donor cells, single-round labeling was used instead of the two-round labeling. Following microarray analysis, Q-PCR of unamplified SCNT and IVF blastocyst cDNA was used to verify the differential expression of the six genes of interest was real and not simply an artifact of the differences in labeling protocols.
Selection of target genes
Initially bovine SCNT and in vivo produced (AI) embryos (three groups of 10 embryos each SCNT and AI) along with several fibroblast donor cell lines were subjected to microarray analysis to measure the degree of reprogramming that occurs between the time of nuclear transfer and the blastocyst stage. Following microarray analysis (Aston et al., 2009a), several genes were selected for further analysis based on degree of change as well as physiological importance. THBS and SNAI2 underwent dramatic downregulation following SCNT and Anxa1 underwent moderate downregulation. Tacstd1 and Oct4 were of interest because they were highly expressed in blastocysts and expressed at low levels or not at all in fibroblast donor cells. Although Tfap2c was not represented on the microarray, this gene was known based on previous work in our laboratory to be unexpressed in fibroblasts and strongly expressed in blastocysts (Aston et al., 2009b).
Reverse transcription and SYBR Green Q-PCR
Reverse transcription was performed using Superscript III Reverse Transcriptase (Invitrogen, Carlsbad, CA) with random primers. Optizyme Recombinant RNase Inhibitor (Fisher Scientific, Fair Lawn, NJ) was utilized at a concentration of 1 unit per μL during the reverse transcription of embryonic RNA. The cDNA was stored at −20°C until use.
SYBR Green real-time PCR (Abgene, Rochester, NY) was used to characterize relative expression levels of Thbs, Snai2, Anxa1, Tacstd1, Oct4, and Tfap2c in fibroblast cells and IVF and SCNT embryos. Expression levels were analyzed by at various stages following fertilization or activation. Following SCNT, embryos 30 min and 12 h postactivation and at the two-cell, four-cell, eight-cell, morula, and blastocyst stages were analyzed. The 30-min postactivation group was analyzed to establish a baseline level of transcript abundance for each gene against which other stages could be compared. An IVF 30-min postfertilization group was not collected because fertilization times can vary following insemination, so the embryos collected would exhibit unacceptable variability in terms of fertilization status and timing (Kim et al., 2002). IVF embryos were analyzed 16 h postinsemination (approximately 12 h postfertilization), and at the two-cell, four-cell, eight-cell, morula, and blastocyst stages. Embryos were pooled in groups of five to provide sufficient RNA for reverse transcription and Q-PCR analysis without the need for linear amplification, and the pooling of embryos served to minimize variability between replicates.
Q-PCR was performed in white thin-walled 96-well plates, and each Q-PCR reaction was performed in triplicate. Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and beta-actin were initially utilized as housekeeping reference genes. In agreement with previously published data, GAPDH was found to be most consistently expressed in preimplantation bovine embryos, and was therefore used as the internal control housekeeping gene (Smith et al., 2007). Primers for Gapdh, Thbs, Snai2, Anxa1, Tacstd1, Oct4, and Tfap2c (Table 1) were designed using Primer3 primer-design software (Rozen and Skaletsky, 2000). All primers utilized were designed and optimized to ensure similar reaction efficiencies between target genes and Gapdh. In addition, cDNA was diluted such that threshold cycles were similar between the target and housekeeping genes. These measures were taken because a critical assumption for the application of the delta-delta Ct method (ΔΔCt) is that the efficiencies of target and housekeeping primers be very similar. Even small variances in primer efficiencies can result in large differences following 30–40 PCR cycles. A standard PCR protocol with a 15-μL reaction volume was used. The reactions consisted of SYBR Green PCR Master Mix containing fluorescein reference dye, forward and reverse primers at 200–300 nM final concentration, and 1-μL diluted template cDNA. The same PCR protocol was used for all primers: 15 min at 95°C for activation of the hot start Thermo-Start DNA Polymerase; 40 cycles of 95°C for 15 sec, 58°C for 30 sec, and 72°C for 15 sec (data collection step), then 95°C for 30 sec followed by an 80-cycle melt curve initiated by 30 sec at 55°C with a temperature increase of 0.5°C each cycle.
Statistical analysis
Microarray
The raw intensity data from the twelve microarray chips were preprocessed together using the RMA algorithm (Irizarry et al., 2003). The limma/eBayes test (Smyth, 2004) was used to test for differential expression between the six donor cell samples and the six embryo samples. The Benjamini-Hochberg adjustment (Benjamini and Hochberg, 1995) was applied to the resulting p-values, and the false discovery rate (FDR) was controlled at 0.01.
Q-PCR
The ΔΔCt method was used for real-time PCR data evaluation (Livak and Schmittgen, 2001). Data was normalized for differing amounts of input cDNA using ΔCt (Ct for the Gapdh housekeeping gene minus Ct for the gene of interest). Next, ΔΔCt was calculated by subtracting the ΔCt of each sample from the ΔCt of a reference liver cDNA sample run in each plate. The n-fold increase or decrease in expression levels of each gene at each embryonic stage was calculated using the formula 2−ΔΔCt. Pair-wise comparisons were performed using the Student's t-test. A probability of p < 0.05 was considered significant.
Results
SCNT and IVF embryo development
In vitro development of IVF and SCNT embryos occurred at comparable rates with approximately 35% fo IVF embryos and 30% of SCNT embryos developing to blastocyst. Rates of development to earlier stages were also similar between groups.
Microarray analysis
After applying the Benjamini-Hochberg adjustment (Benjamini and Hochberg, 1995) to the p-values resulting from the comparison of chips from six donor cell lines with six embryo chips (three SCNT and three AI) using the limma/eBayes test (Smyth, 2004) for differential expression and controlling the FDR at 0.01, there were 10,942 probe sets (out of 24,128) called significantly differentially expressed between fibroblast donor cells and all embryos. Figure 1 is a volcano plot of the fibroblast/embryo comparison. Figure 2 is a heatmap of five of the genes selected for further analysis. Tfap2c was not represented on the microarray chip.
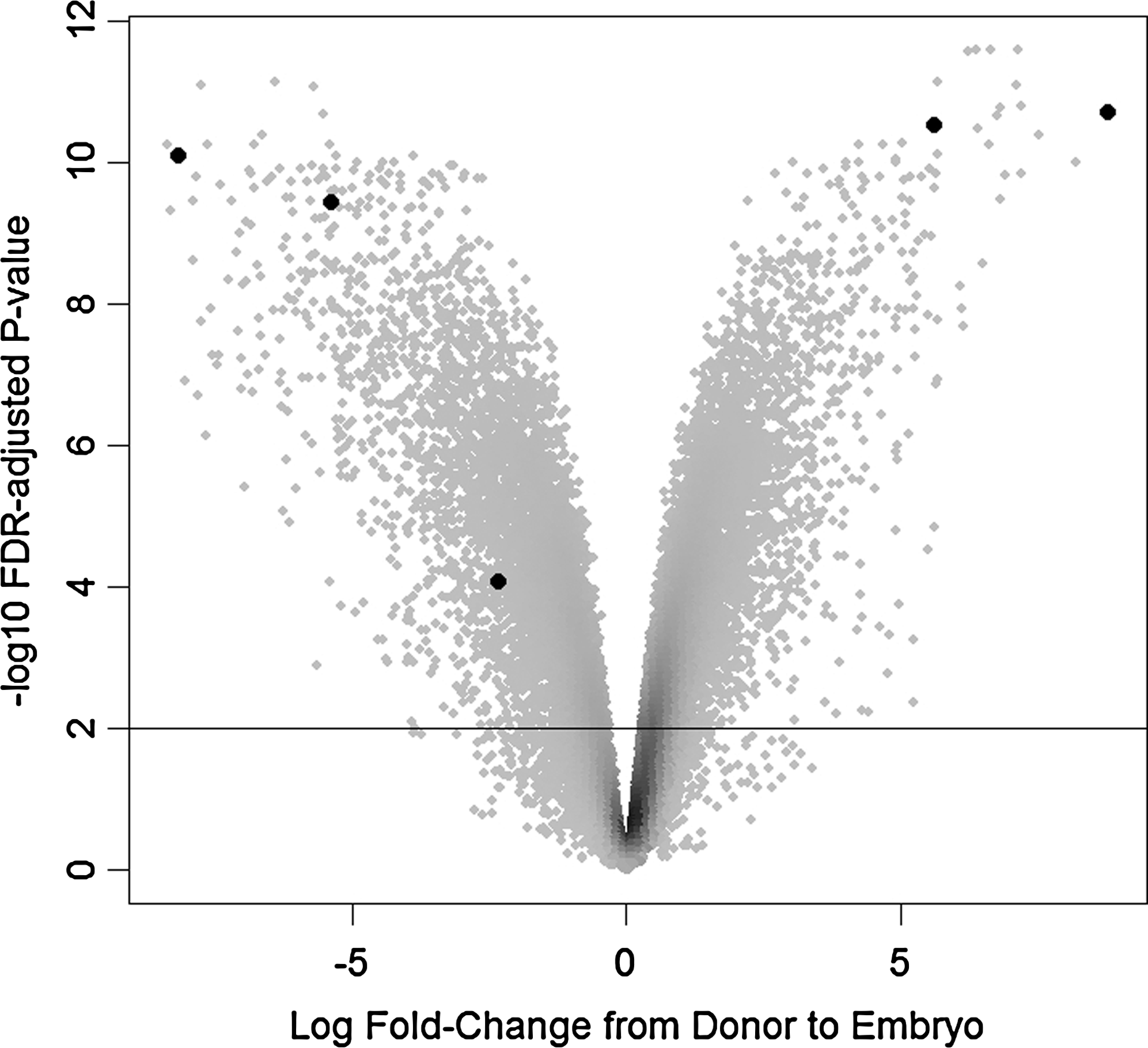
Volcano plot summarizing results, with points colored by density. Points above the reference line correspond to probe sets called significant when controlling the FDR at 0.01. Five of the genes selected for further evaluation are highlighted. The highlighted points are, left to right: Thbs, Snai2, Anxa1, Oct4, and Tacstd1.

Heatmap of five genes of interest. The color scale is from dark for low expression values to light for high expression values. The columns are labeled “E” for embryo and “D” for donor cell samples.
Q-PCR analysis
The transcript abundance of Thbs, Snai2, Anxa1, Tacstd1, Oct4, and Tfap2c were analyzed by Q-PCR in various stages of SCNT and IVF embryos as well as donor cells. For every gene except Tacstd1 there were differences in transcript levels between SCNT and IVF embryos in multiple embryonic stages. Interestingly, these differences were generally rectified by the blastocyst stage so SCNT and IVF embryos were indistinguishable at the blastocyst stage for every gene except Oct4, which was present at a lower level in SCNT embryos at every stage of development. The results of the Q-PCR experiments are represented graphically in Figure 3.

Relative expression of genes based on Q-PCR. White and black bars represent SCNT and IVF embryos, respectively. Checkered bars represent donor cells and oocytes. Lowercase superscripts compare SCNT embryos, and uppercase superscripts compare IVF embryos between stages. Stages with unlike superscripts are different (p < 0.05). Asterisk indicates expression levels between SCNT and IVF embryos of the same stage differ (p < 0.05). The stage with highest expression for each gene is scaled to 1.0 on the y-axis, and other stages are scaled accordingly. Abbreviations are: Fibroblast (Fib), Oocyte (Oo), 30-min postfusion (30 m), 12 h postfusion/fertilization (12h), two-cell (2c), four-cell (4c), eight-cell (8c), Morula (Mor), Blastocyst (Bl).
Thbs, Anxa1, and Snai2 exhibit similar patterns of gene regulation following SCNT, although the timing differs for each gene. In each case transcript level remains higher in SCNT embryos than IVF embryos through several cell cycles: through the four-cell stage for Thsb1, through the eight-cell stage for Anxa1, and through the morula stage for Snai2. In the case of Thsb1, transcript abundance is the same between SCNT and IVF embryos at the eight-cell, morula, and blastocyst stages, Anxa1 transcript level is equivalent at morula and blastocyst, and Snai2 transcript remains higher in SCNT embryos until the blastocyst stage when it is abruptly shut off. Tacstd1, Tfap2c, and Oct4 all require transcriptional induction as embryos develop to blastocyst. In general patterns of transcript abudance in SCNT embryos closely resemble those of IVF controls with some important differences. Levels of Tacstd1 do not differ between SCNT and IVF embryos at any stage; however, Tfap2c and Oct4 both exhibit differential transcript levels. Tfap2c is detectable at the eight-cell stage in SCNT embryos but not until morula in IVF embryos, and although expression appears to be induced earlier in SCNT embryos it is underrepresented in SCNT morulae. Tfap2c transcript level declines significantly between morula and blastocyst stages in IVF embryos, and declines to a lesser degree in SCNT morulae so that levels are equivalent at the blastocyst stage. Of the six genes analyzed, OCT 4 is the only one differentially represented at the blastocyst stage. In fact, Oct4 expression is significantly higher in IVF embryos at every stage analyzed except in four-cell embryos where the difference approaches significance (p = 0.082).
Discussion
The comparable rates of embryo development to blastocyst are one indication that nuclear reprogramming is relatively efficient following SCNT and that sufficient reprogramming has occurred in the early embryo to direct early embryonic cell differentiation.
In agreement with the in vitro development data, the microarray studies indicate that a substantial amount of reprogramming of the donor cell genome has occurred by the blastocyst stage. Microarray analysis of donor cell expression patterns compared with SCNT and AI blastocysts combined found differential expression of 10,942 probe sets. Remarkably, by the blastocyst stage, a similar analysis comparing SCNT and AI blastocysts found only 28 probe sets differentially expressed (Aston et al., 2009a). These results are quite similar to previously published results (Smith et al., 2005). Despite the apparent efficiency with which the somatic cell genome is reprogrammed by the blastocyst stage, of the six genes analyzed by Q-PCR, five were differentially represented at two or more stages prior to blastocyst formation. These results indicate a substantial amount of time is required for the SCNT transcript profile to “catch up” with the profile of control embryos. This is not surprising given the fact that the oocyte is designed to reprogram gamete nuclei, and the epigenetic modifications to somatic cells are much different than those of germ cells. The remarkable thing is the adaptability of the oocyte cytoplasm to successfully reprogram a variety of different somatic cell types, albeit inefficiently. A recent study evaluating the global transcriptome of murine SCNT embryos during the first two cell cycles also indicated a large degree of aberrant transcript levels in early mouse SCNT embryos. It was also found that transcription of the donor cell genome continues during the first cell cycle when the embryonic genome is typically silenced (Vassena et al., 2007).
The genes analyzed by Q-PCR were selected because they exhibited dynamic changes in transcript abundance in SCNT embryos and because of their important biological functions in early development and differentiation. OCT4 is a homeodomain transcription factor and a hallmark of undifferentiated stem cells. Reduced expression of Oct4 has been shown in mouse embryos (Niwa et al., 2000) and human embryonic stem cells (Matin et al., 2004) to result in trophoblast differentiation. Conversely, overexpression of the gene results in differentiation of primitive endoderm and mesoderm (Niwa et al., 2000). Precise expression levels of Oct4 are clearly important in proper early embryonic development. SNAI2 is a member of the Snail family of transcription factors that also has important roles in early development (Cobaleda et al., 2007). It has been shown to be required for gastrulation, epithelial–mesenchymal transition, and cell survival in the mouse (Sefton et al., 1998). TACSTD1 is believed to be important in directing cell migration during early development in zebrafish (Villablanca et al., 2006). TFAP2C is a transcription factor that appears to be an important regulator of trophoblast development and differentiation (Li and Kellems, 2003). Tfap2c-null mice die around embryonic day 7.5 as a result of malformation of extra-embryonic membranes (Auman et al., 2002; Winger et al., 2006). THBS is a secreted glycoprotein involved in cell migration and proliferation and is apparently important in ossification, and neural and lung development (Iruela-Arispe et al., 1993). ANXA1 is a calcium and phospholipid binding protein that has been shown to be involved in membrane trafficking, cell division, and differentiation. Annexins are expressed in a broad range of tissue types, possibly indicating they play important roles in basic cell physiology (Gerke and Moss, 2002).
Each of the genes analyzed is functionally important during early development, and even transient differences in transcript abundance could potentially have negative consequences downstream. Complete nuclear reprogramming following SCNT would result in expression profiles in SCNT embryos that mirror IVF profiles. This is not the case for any of the genes analyzed except for Tacstd1. Quantitative analysis of these genes only at the blastocyst stage would indicate, with the exception of Oct4, that proper reprogramming has occurred. Studies that evaluate gene expression only at the blastocyst stage might underestimate the scope of transcriptome abberations following SCNT. The importance of the early embryonic transcript levels of the genes analyzed in this study remains to be seen, but it is probable that any divergence from normal transcript levels at any stage of development and for any amount of time has a negative effect on the health of the embryo. The fact that Tacstd1 was present at normal levels in SCNT embryos indicates that some genes may be more amenable to reprogramming following SCNT than others. Understanding the properties of genes that make them more reprogrammable might offer insights into nuclear reprogramming mechanisms.
Thbs, Snai2, and Anxa1 were all highly abundant transcripts in fibroblast donor cells, and consequently, transcript persisted at an above-normal level in early SCNT embryos. This observation is in agreement with previous reports of ectopic expression of fibroblast-specific genes following SCNT (Ng and Gurdon, 2005); however, it is not clear based on the current study whether the abnormal persistence of transcript resulted from the persistence of gene transcription or whether the transcripts detected were residual from the donor cell. Further research will be important to characterize the mechanisms involved in donor cell-specific transcript clearance from SCNT embryos as well as the effect of residual transcript on embryonic and fetal development. Following SCNT 80–90% of nonhistone proteins are removed from somatic nuclei effectually erasing the somatic cell transcription program (Gurdon et al., 1979). This is followed by reestablishment of an embryonic transcription program by numerous chromatin factors (CFs) (Gao et al., 2007). Incomplete erasure of epigenetic modifications prior to CF reestablishment is a potential cause of overexpression. Likewise, the differential levels of transcript abundance for Tfap2c and Oct4 might be caused by similar deficiencies in the process of epigenetic erasure or the subsequent process of epigenetic reestablishment (Aston et al., 2009a; Gao, et al., 2007).
The present study further characterizes the deficiencies associated with appropriate transcriptome levels following SCNT. In addition to aberrant gene expression (Arnold et al., 2006; Herath et al., 2006; Hill et al., 2002; Humpherys et al., 2002; Niemann, et al., 2002; Pfister-Genskow et al., 2005) and incomplete or inefficient epigenetic modification (Alberio and Campbell, 2003; Cezar et al., 2003; Enright et al., 2003; Kang et al., 2001; Kremenskoy et al., 2006; Santos et al., 2003) following SCNT, it is apparent from this study as well as the work by Vassena et al. (2007) that the earliest embryonic stages in SCNT embryos are highly divergent from control embryos in terms of transcriptional profiles. The fact that these highly aberrant transcriptional profiles can be almost completely rectified by the blastocyst stage following SCNT and a portion of these embryos have the capacity to develop normally to term reflects the incredible plasticity of the oocyte in its reprogramming activities. Great strides have been made in understanding the molecular mechanisms associated with nuclear reprogramming following SCNT, and continued progress will ultimately lead to improved SCNT efficiency as well as increased understanding of universal epigenetic mechanisms associated with cancer and stem cell biology as well as early development.
Footnotes
Acknowledgments
This work was funded in part through a grant from the USDA CREES (08-03183) to K.L.W., and is published as Utah Agricultural Experiment Station publication No. 8045.
Author Disclosure Statement
The authors declare that no comflicting financinal interests exist.