
Abstract
Abstract
Embryonic stem cells (ESCs) have the potential to reprogram somatic cells into ESC-like cells through cell fusion. In the present study, the potential of human (h)ESC cytoplasts and karyoplasts to reprogram human hepatocytes was evaluated. Green fluorescent protein (GFP) transfected hESCs (ENVY cells) were fused with SNARF-1 (CellTracker)-labeled human hepatocytes using polyethylene glycol (PEG) and fluorescence-activated cell sorting (FACS) to produce hESC-hepatocyte hybrids. Immunocytochemical analysis of ESC markers showed that the hybrids expressed OCT4, TRA-1-60, TRA-1-81, SSEA-4, and GCTM-2. However, SSEA-1, which is typically low or absent on hESCs, was detected on hESC–hepatocyte hybrids. Moreover, reverse transcriptase polymerase chain reaction (RT-PCR) showed that alpha-fetoprotein, which is highly expressed in hepatocytes, was erased in the hybrids. These results indicated that hESCs have the potential to reprogram hepatocyte phenotype to a relatively undifferentiated state, but such hybrid cells are not identical to hESCs. Although hESC–hepatocyte hybrids were aneuploid, they were able to differentiate into embryoid bodies and some types of somatic cells. Furthermore, cybrids of enucleated hESCs and hepatocytes were produced by cell fusion, but the cybrids were unable to self-renew in the same way as hESCs. Presumably, the reprogramming factors are associated with the karyoplast and not the cytoplast of hESCs.
Introduction
For human cell replacement therapy (CRT), development of patient specific ESC-like pluripotent cells may be needed to avoid immune rejection in allogeneic stem cell transplantation. Importantly, personalized stem cells can be studied in vitro to understand and to attempt correction of any identified genetic defect by modification of the personalized stem cells. A range of strategies to generate patient-derived pluripotent cells, such as somatic cell nuclear transfer (SCNT) (French et al., 2006), cell fusion (Pralong et al., 2006), and induced pluripotent stem (iPS) cells by viral transduction using transcription factors (Jaenisch and Young, 2008), have been attempted. So far, SCNT-derived ESCs have only been reported in mice (Wakayama et al., 2001) and nonhuman primates (Byrne et al., 2007). Although human cloned blastocysts were obtained from SCNT (French et al., 2008), SCNT-derived ESC lines have not yet reported. iPS cells, believed to be similar to ESCs in pluripotency, have been generated from fibroblasts (Nakagawa et al., 2008; Park et al., 2008; Yu et al., 2007) by genetic modifications and using recombinant proteins (Zhou et al., 2009). However, the viral transfection systems used to insert the genes at random locations in the cells is a major safety concern for iPS cells in regenerative medicine. New strategies will need to be developed for clinical purposes to avoid potential harm from genetic modification or products from bacteria.
The reprogramming potential of human (h)ESC has been demonstrated through reprogramming adult somatic cell into the undifferentiated state by cell fusion (Cowan et al., 2005). This would enable stem cells to be created by fusion with hESCs. Although cell reprogramming by cell fusion was achieved from a range of somatic cells and protocols in mice (Do and Scholer, 2005; Matsumura et al., 2007; Pralong et al., 2005; Silva et al., 2006) and in humans (Yu et al., 2006), it has been stated that the safety issues regarding the generation of tetraploid cells for potential clinical therapies, but which may be potentially cancer forming in the patient, need to be seriously considered (Sullivan and Eggan, 2006). Diploid cybrid cells that are reprogrammed by fusion of hESC cytoplasm and somatic cells have also been reported (Strelchenko et al., 2006). If hESC cytoplasts were able to reprogram somatic cells, it would be a significant advance for CRT to avoid the problems associated with the need to access large numbers of human oocytes for reprogramming. Generation of reprogrammed diploid cells for CRT by cell fusion still remains to be investigated because of the risk of tetraploidy or polyploidy. Thus, the reprogramming potential of hESC cytoplasts needs to be confirmed.
Type 1 diabetes occurs in genetically predisposed individuals when the immune system attacks and destroys specifically the insulin-producing β-cells of the pancreatic islets. The only treatment for type 1 diabetes is replacement of the β-cell mass by ectopancreatic transplant or pancreatic islet implant. Liver and pancreas arise from common endodermal progenitor cells (Grompe, 2003; McLin and Zorn, 2003), and purified hepatic oval stem cells or fetal liver stem/progenitor cells possess the capacity to trans-differentiate into functional endocrine cells, including insulin-producing cells under some specific culture conditions (Feng et al., 2005; Horb et al., 2003; McLin and Zorn, 2003; Yang et al., 2002; Zhou et al., 2008); thus, hepatocytes and β-cells are considered to be end-stage differentiated cells and are unable to be propagated in culture. Efforts to produce islet neogenesis or to initiate β-islet growth in vitro from either fetal or adult tissue have had minimal success. To reverse hepatocytes into stem/progenitor cells will be the only option to generate insulin-producing β-cells. Indeed, hepatocyte hybrids have been reported by cell fusion from the monkey embryoid body (Okamura et al., 2006), but no attempts have been made to generate human hepatocyte hybrids and cybrids.
In the present study, the cellular reprogramming potential of hESC cytoplasts and karyoplasts was investigated using cell fusion. Binucleated hepatocytes are commonly found in the healthy human liver and are formed during normal development (Kudryavtsev et al., 1993; Seglen, 1997). Therefore, it should be possible to generate polyploid hybrids from human hepatocytes without gene transfection and drug selection to satisfy the requirements for CRT.
Materials and Methods
All chemicals were purchased from Sigma-Aldrich Pty. Ltd (Castle Hill, NSW, Australia) unless otherwise stated. Cell cultureware were from BD Biosciences (North Ryde, NSW, Australia), and somatic cells were cultured in BD Falcon cell culture flask unless otherwise indicated. All GIBCO solutions were purchased from Invitrogen Australia Pty Ltd, Mount Waverley, VIC, Australia.
Human somatic cell culture
Cryopreserved human primary hepatocytes (hNHeps; CC-2591, Lot number 5F0063, Cambrex Bio Science, Walkersville, MD, USA), which was isolated from a 3-day female, were thawed according to the manufacturer's instructions. Briefly, the hepatocytes were thawed in a 37°C water bath with gentle shaking and the cell suspension was slowly transferred into 20 mL of cold hepatocyte culture medium (HCM; CC-3198, Cambrex Bio Science). After centrifugation at 50 × g at 4°C for 3 min, the cell pellet was suspended in HCM and seeded on BD biocoat collagen I 25 cm2 flasks to give a concentration of 1.2 × 105 cells/cm2. Hepatocytes were incubated in HCM at 37°C in 5% CO2 in air with 95% relative humidity and passaged every 7 days. The cell culture medium was replaced by minimum essential medium alpha (α-MEM; Gibco, SKU# 12561-056) supplemented with 10% fetal bovine serum (FBS; Gibco, SKU# 10099-141) after two passages. The culture medium was changed every 3 to 4 days. The cells were cryopreserved in a solution containing 90% FBS plus 10% dimethyl sulfoxide (DMSO) and stored in liquid nitrogen. Before cell fusion, the hepatocytes were thawed in α-MEM supplemented with 10% FBS.
Human skin fibroblasts (CCD-919Sk; CRL-1826, ATCC, Manassas, VA, USA) and human foreskin fibroblasts (HFF-1; SCRC-1041, ATCC) were maintained in α-MEM supplemented with 10% FBS. CCD-919Sk cells at passage 10 were cryopreserved as described above. Before cell fusion, CCD-919Sk cells were thawed in α-MEM supplemented with 10% FBS. HFF-1 cells at passage 25 were dissociated by TrypLE Select (Gibco, SKU# 12563-029) and suspended at 1.0 × 106 cells/mL in 10 ml Ca/Mg-free Dulbecco's phosphate-buffered saline (D-PBS; Gibco, SKU# 14190-144) and irradiated by 6500 rads of γ-irradiation with Gammacell-1000 (MDS Nordion, Kanata, Ontario Canada). γ-irradiated cells were then cryopreserved until they were used as feeder cells for culture of hESCs and their hybrid and cybrid cells.
Human ESCl culture
A green fluorescent protein (GFP) transfected-hESC line (ENVY cells) (Costa et al., 2005) was cultured on BD Falcon 60-mm center-well organ culture dishes on γ-irradiated HFF-1 feeder cells in Knockout-Dulbecco's modified Eagle's medium (KODMEM; Gibco, SKU# 10829-018) supplemented with 20% Knockout-Serum Replacement (KOSR; Gibco, SKU# 10828-028), 2 mM L-glutamine (Gibco, SKU# 25030-081), 1% MEM nonessential amino acids solution (NEAA; SKU#11140-050), 1% Insulin-Transferrin-Selenium (ITS; Gibco, SKU# 51500-056), 0.1 mM 2-Mercaptoethanol (Gibco, SKU# 21985-023), 4 ng/mL recombinant human FGF basic (hbFGF; 233-FB/CF, R&D Systems, Inc., Minneapolis, MN, USA), and 1% penicillin–streptomycin (Pen/Strep; Gibco, SKU# 15070-063). hESC colonies were mechanically dissociated into small clumps at 6- to 8-day intervals with a fine needle and transferred onto fresh feeder cells for passage. Only undifferentiated cells judged by morphology were passaged.
Bulk culture of hESC cells was carried out according to a previously described method (Suemori et al., 2006). Briefly, the hESCs were dissociated by culture in TrypLE Select for 5 min at 37°C, and the dissociated cells were filtered through a 100-μm cell strainer (BD Biosciences). After washing with KODMEM, the cells were plated onto 25 cm2 culture flask containing γ-irradiated HFF-1 feeder cells. Bulk cultured hESCs were passaged every 3 to 5 days. The culture medium was replaced every day following 48 h of initial passage.
Generation of hESC hybrids
hNHeps (at passage 3–5) cells were washed with D-PBS and incubated in D-PBS containing 20 μM seminaphthorhodafluor-1-acetoxymethyl ester (SNARF-1; S22801, Molecular Probes Invitrogen, Carlsbad, CA, USA) for 30 min to label the cells. After staining, the cells were washed and cultured with 15 mL α-MEM plus 10% FBS at 37°C for at least 30 min. For cell fusion, SNARF-1-labeled hNHeps and bulk cultured ENVY (colony culture for 96 passages and bulk culture for 24–25 passages) cells were detached from flasks by TrypLE Select. The single-cell suspensions were isolated by gently pipetting and filtering through 40-μm cell strainers (BD Biosciences). Cell fusion was performed in 50-mL conical tubes. Briefly, hNHeps and ENVY cells were mixed (1:1 at 1 × 106 cells/mL) in D-PBS by gently pipetting. The mixture was centrifuged at 130 relative centrifugal force (RCF) for 5 min and the supernatant was removed. The cell fusion was carried out in 0.5 mL prewarmed D-PBS containing 42% polyethylene glycol 1500 (PEG) and 5% DMSO (PEG/DMSO) for 1 min. An additional 10 mL of KODMEM plus 20% KOSR was added and the cell suspension was stirred for 5 min constantly. After centrifugation at 130 RCF for 5 min, PEG/DMSO was removed and the cell pellet was resuspended in 1 mL of hESC culture medium and seeded onto a 75-cm2 flask containing γ-irradiated HFF-1 feeder cells. After culture for 72 h, the cells were detached by TrypLE Select. Single cells were analyzed with FACSVantageSE-Diva (BD Biosciences) after filtering through a 40-μm cell strainer. The fused cells that were positive for both GFP and SNARF-1 were collected into 1 mL of hESC culture medium and plated on a 60-mm center-well organ culture dish containing γ-irradiated HFF-1 feeder cells. The culture medium was changed at 48 h of culture and then every day thereafter. The ENVY hybrids were passaged every 5 to 7 days. CCD-919Sk cells (at passages 10–11) fused with ENVY cells in the same way as for hNHeps were used as a control.
Immunocytochemistry of hESC hybrids
Colony cultured ENVY (at passage 97) and ENVY-hNHeps hybrid (at passage 7) cells were cut into small pieces and transferred onto sterile 12-well glass slides (Cat. No. 6041205, MP Biomedicals Australia, Seven Hills, NSW, Australia) containing γ-irradiated HFF-1 feeder cells in hESC culture medium. After culture for 5 to 7 days, the colonies were fixed with 100% cold ethanol (−20°C) for 10 min. The ethanol was removed and the slide was air dried for 30 min at room temperature. After washing with D-PBS, the cells in the wells of the slide were incubated with human-specific antibodies for transcription factor OCT4 (1:20; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), keratan sulfate-associated antigen TRA-1-60 (1:20; MAB 4360, Chemicon International Inc., Tumecula, CA, USA), TRA-1-81 (1:20; MAB4381, Chemicon International Inc.) and GCTM-2 (provided by Dr. Martin Pera, Monash Institute of Medical Research, Australia), stage-specific embryonic antigen (SSEA)-1 (1:20; MAB4301, Chemicon International Inc.) and SSEA-4 (1:20; MAB4304 Chemicon International Inc.) at room temperature for 30 min, respectively. After washing, the cells were incubated for 30 min at room temperature with goat antimouse IgG conjugated with Alexa Fluor 647 (1:40; A21235, Molecular Probes Invitrogen) for OCT4, SSEA-4 and GCTM-2 and incubated with goat antimouse IgM conjugated with Alexa Fluor 647 (1:40; A21238, Molecular Probes Invitrogen) for TRA-1-60, TRA-1-81, and SSEA-1, respectively. ENVY cells incubated with goat antimouse IgG or goat antimouse IgM were employed as negative controls. After immunostaining, the cells were incubated with D-PBS containing 20 μg/mL Hoechst 33342 for 5 min. The slides were then washed and mounted using Vectashield mounting medium (Vector Laboratories Inc., Burlingame, CA, USA) to prevent loss of fluorescence during examination. The slides were covered by coverslips and permanently sealed around the perimeters for prolonged storage. All fluorescence images were acquired and analyzed using an Olympus Fluoview 1000 confocal microscope and FV10-ASW software (version 1.3c, Olympus).
RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR) of hESC hybrids
Total RNA was extracted from hNHeps (at passage 6), CCD-919Sk (at passage 12), hESC (at passage 108), and ENVY-hNHeps hybrid cells (at passage 8) using the RNeasy minikit (Qiagen, Chatsworth, CA, USA) according to the instructions of the manufacturer. A total of up to four dishes of ENVY and ENVY hybrid cell colonies were detached from the dishes with a fine blunt-end glass pipette and transferred into RNase-free polypropylene centrifuge tubes for RNA extraction. The RNAs were quantified by NanoDrop ND-1000 (NanoDrop products, Wilmington, DE, USA) and stored at −80°C until use. cDNA was synthesized from 1–4 μg of total RNA using the cDNA synthesis system (Superscript III First-Strand Synthesis System; Invitrogen) with 50 μM Oligo(dT) 20 following the manufacturer's protocol. The reaction was performed at 55°C for 50 min. After incubation at 85°C for 5 min to terminate the reaction, 1 μL of RNase H was added into the tube and incubated at 37°C for 20 min. cDNA products were stored at −20°C or used for PCR immediately. RT-PCR was performed according to the manufacturer's instructions from Platinum Taq DNA Polymerase High Fidelity (SKU# 11304-011, Invitrogen) in 50 μL of PCR Buffer containing 0.2 μL (1.0 U) of Platinum Taq High Fidelity, 2.5 μL of each primer of human transcription factor (hOCT4), human alpha-fetoprotein (hAFP) and human β-actin (hACT), 2.0 μL of cDNA from each sample. The PCR conditions for amplification involved an initial 5 min denaturation step at 94°C, followed by 30 cycles, each consisting of a 30-sec denaturation step at 94°C, a 30-sec annealing step at 60°C, and a 1-min extension step at 72°C. The reactions were terminated by a final elongation step of 5 min at 72°C. The PCR products were visualized on a 2.0% (w/v) agarose gel stained with ethidium bromide and photographed under Bio-Rad Gel Doc XR (Bio-Rad Laboratories, Hercules, CA, USA). The 50-bp DNA ladder (New England Biolabs, Ipswich, MA, USA) was used as a molecular marker. The primer sequences used for PCR products are shown in Table 1.
Karyotype analysis of hESC hybrids
Karyotyping of ENVY–hNHeps hybrids at passage 3 was performed by Southern Cross Pathology Australia. Numbers of chromosomes were counted using an Olympus IX81 microscope equipped with a 60 × water objective.
In vitro differentiation of hESC hybrids
Embryoid body formation of ENVY hybrids was carried out at passage 7. Briefly, the hybrid cells were detached by gentle scraping and pipetting and plated onto nontreated polystyrene Petri dishes in hESC culture medium without ITS and hbFGF. The medium was changed every 5 days. After 10 days of culture, the embryoid bodies were further cultured in α-MEM supplemented with 10% FBS in Falcon six-well cell culture plates (Cat. No. 353046; BD Biosciences). Spontaneous differentiation of the hybrid cells was evaluated 10 days later.
Generation of hESC cybrids
Conditioned medium (CM) was produced from HFF-1 cultures as previously described. Briefly, γ-irradiated HFF-1 cells were cultured in hESC culture medium, and CM was harvested every day for 6 days from the second day of culture. The CM was filtered through a 22-μm filter and stored at −20°C. CM was thawed and supplemented with 4 μg/mL hbFGF just before use.
ENVY cell colonies were detached from the culture dish by gently scraping with a fine blunt-end glass pipette, and then were disassociated by gentle pipetting. The disassociated cells were filtered through a 70-μm cell strainer, centrifuged, and resuspended in 2 mL CM. The cell suspension was plated (0.5 mL) onto 22 μm Thermanox plastic coverslips (NUNC; Cat. No. 174977, Thermo Fisher Scientific, Rochester, NY, USA). These coverslips were preincubated with diluted BD Matrigel matrix (1:30; 354234, BD Biosciences) at room temperature (22°C) for at least 1 h before cell plating. The cells were cultured overnight on coverslips at 37°C in 5% CO2 in air with 95% relative humidity. At 24 h of culture, the culture medium was replaced by enucleation medium and the cells were further cultured in enucleation medium for 1–2 h. Enucleation medium consisted of 400 μL of CM supplemented with 1.5 μg/mL cytochalasin D and 1 μg/mL nocodazole. The enucleation was performed at 35°C by centrifugation using a Sorvall Evolution RC centrifuge (Thermo Fisher Scientific Inc., Waltham, MA, USA) with an ss-34 rotor. Briefly, the coverslips were inverted, placed into 50- mL polycarbonate centrifuge tubes (Nalgene; Cat. No. 3117-0500, Nalge Nunc International, Rochester, NY, USA) on centrifugation medium (hESC culture medium containing 6.6%, 8.0%, 10%, 12.5% or 15% Ficoll-400 supplemented with 10 μg/mL cytochalasin D and 3 μg/mL nocodazole) and centrifuged at 25,000 RCF for 50 min; or centrifuged for 50 min at 20,000, 25,000, 30,000, or 35,000 RCF in centrifugation medium containing 10% Ficoll-400 supplemented with 10 μg/mL cytochalasin D and 3 μg/mL nocodazole. After centrifugation, the coverslips were washed and cultured in hESC culture medium overnight. The enucleation was verified by staining the cells with 5 μg/mL Hoechst 33342. Enucleated hESCs were detached by TrypLE Select from four coverslips and TrypLE Select was removed by centrifugation. The cell pellets were combined into 1 mL and mixed with CCD-919Sk cells in D-PBS at a ratio of 1:2. The cell fusion was carried out similarly to that used for generation of hybrids but in 200 μL of D-PBS containing PEG/DMSO. The fused cells were plated onto two 60-mm center-well organ culture dishes containing γ-irradiated HFF-1 feeder cells. The culture medium was changed every day after 72 h of initial culture.
Statistical analyses
Bulk enucleation of hESCs by centrifugation on different conditions was analyzed by chi-square. A p-value <0.05 was considered to be statistically significant.
Results
Generation of hESC hybrids
SNARF-1 loaded human fibroblasts (Fig. 1A and B) and hepatocytes (Fig. 1G and H) were visualized under a fluorescence microscope and positively stained cells were used for cell fusion. After fusion, hybrid cells presenting both GFP (green) and SNARF-1 (red) were 0.8% of the cells for hESC-fibroblast (ENVY-CCD919SK) (Fig. 1C) and 0.9% for hESC-hepatocyte (ENVY-hNHeps) hybrids (Fig. 1I); leakage control was less than 0.1% (Fig. 1F). Enough hybrid cells were collected for further culture, despite the low fusion efficiency. Such hybrid cells formed ESC-like colonies by day 7 of culture (Fig. 1D and E, J and M). These hybrid cell colonies were passaged similar to hESCs (Fig. 1K and N, L and O), and hybrids at least eight passages in culture without morphological changes were used for RNA extraction.
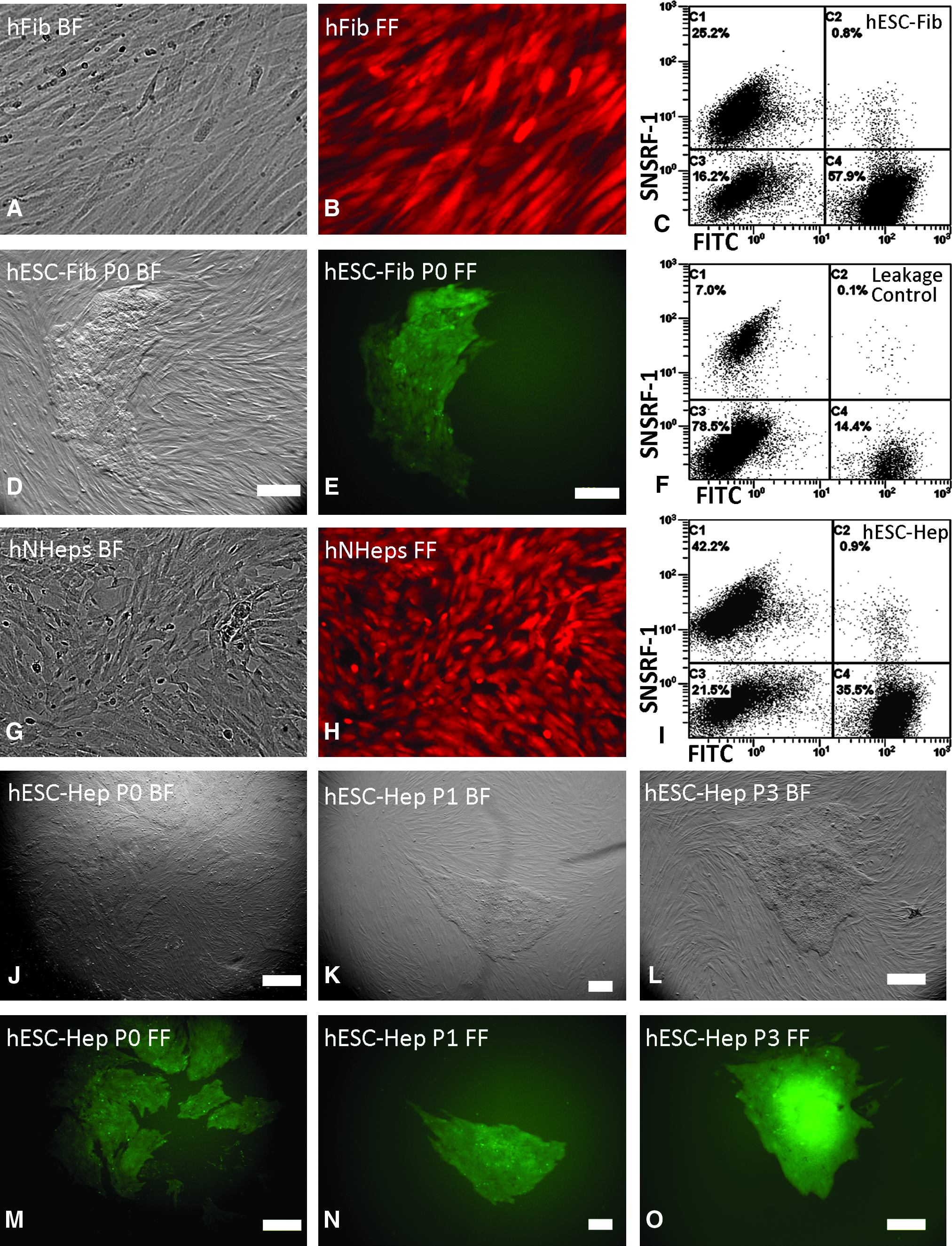
Generation of hESC hybrids by cell fusion of human somatic cells and hESCs. Human skin fibroblasts, CCD-919Sk cells, and human hepatocytes, hNHeps cells loaded with SNARF-1 are shown under bright-field (
Immunocytochemistry of hESC hybrids
Immunocytochemistry showed that the hESC hybrids derived from human fibroblasts and hepatocytes expressed pluripotent stem cell markers, although the expression of pluripotency markers was highest and most consistent at the edge of the colony. Figure 2 showes that hESC–hepatocyte hybrid cells were positively stained for OCT4, TRA-1-60, TRA-1-81, SSEA-4, and GCTM-2 (Fig. 2). The marker SSEA-1 was undetected on hESC–fibroblast hybrids and hESC (SSEA-1 negative staining is not shown), but it was weakly expressed on hESC–hepatocyte hybrids (Fig. 2). hESCs (ENVY) stained for OCT4 were used as positive controls and stained with goat antimouse IgG were employed as negative controls (Fig. 2).
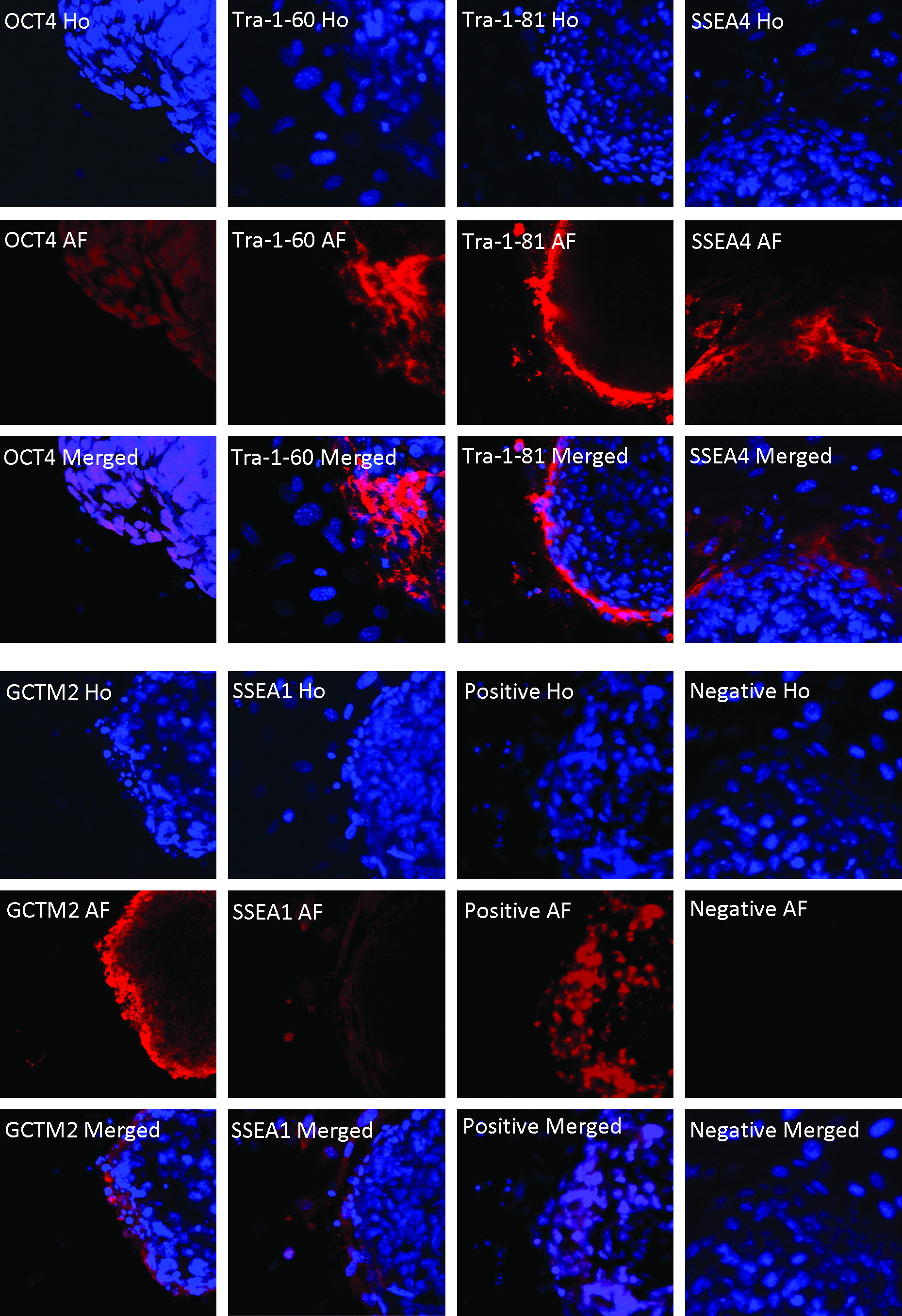
Confocal images of immunochemistry of hESC hybrids under Olympus Fluoview 1000 confocal microscope (original magnification ×60). The images were taken from DAPI (Ho), Alexa Fluor 647 (AF), and merged Alexa Fluor 647 fluorescent into DAPI (Merged) for ENVY–hNHeps hybrid cells. ENVY–hNHeps hybrid cells that were stained positive for OCT4, Tra-1-60, Tra-1-81, SSEA-4, GCTM-2, and SSEA-1 are shown. hESCs, ENVY cells, stained for OCT4 are shown as positive controls (Positive); ENVY cells stained with goat antimouse IgG were used as negative controls (Negative).
RT-PCR and karyotyping of hESC hybrids
Expression of OCT4 was confirmed in hESC–hepatocyte and hESC–fibroblast hybrids by RT-PCR (Fig. 3A). hAFP, an endoderm cell marker, was positive for fetal hepatocytes; however, it was turned off in hESC–hepatocyte hybrids (Fig. 3B).
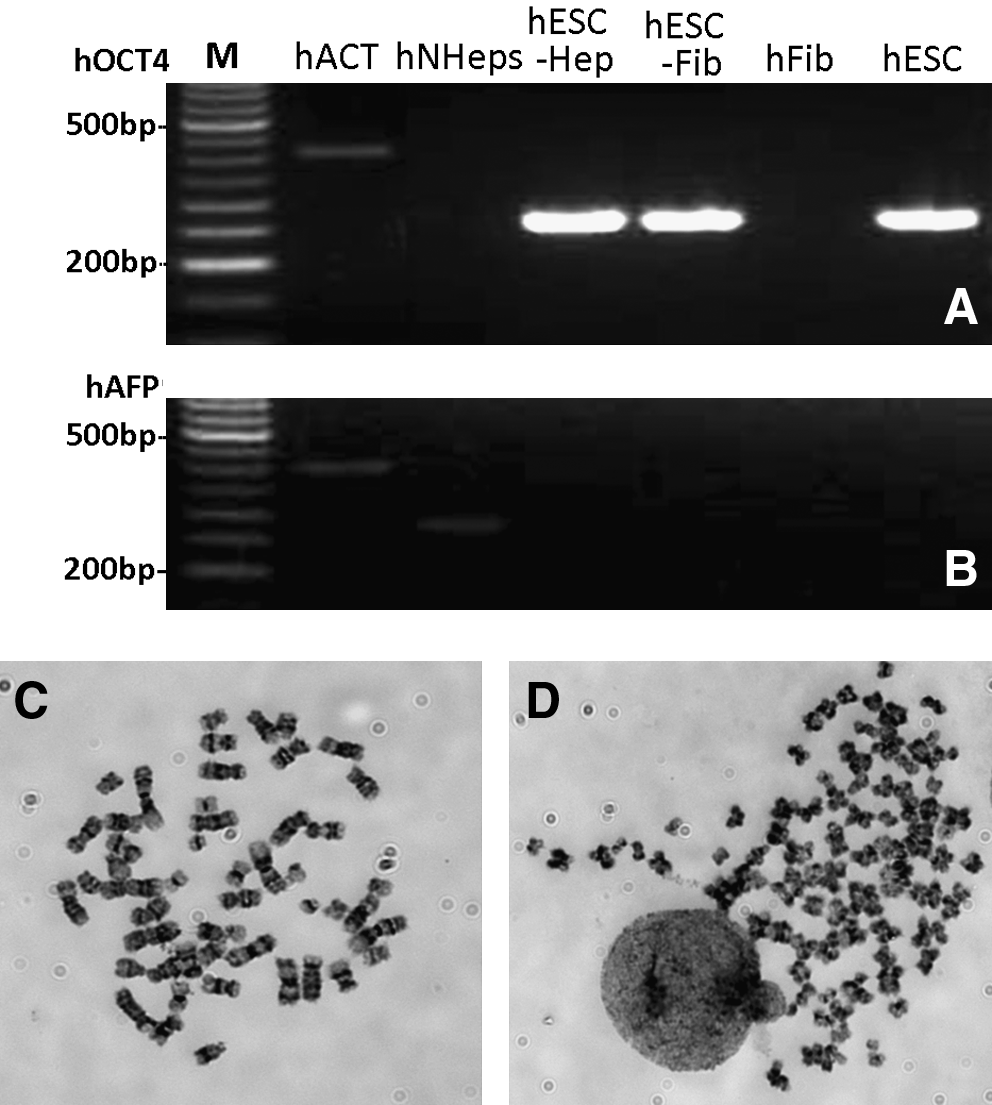
RT-PCR and karotype of hESC hybrids. RT-PCR products of human hepatocytes, fibroblasts and ENVY–hNHeps hybrid cells were detected by 2.0% agar electrophoresis for hOCT4 (
Karyotype of hESC–hepatocyte hybrids at passage 3 showed that the hybrid cell line contained both near-tetraploid and near-diploid chromosome complements with a higher variability in the number of chromosomes. Out of 88 cell spreads from the hybrid cells, 76.1% (67/88) of them were close to diploid chromosomes (Fig. 3C), whereas 23.9% of the cells were close to tetraploid chromosomes (Fig. 3D).
Formation of embryoid bodies and in vitro differentiation of hESC hybrids
Differentiation potential of the hESC–hepatocyte was demonstrated by formation of embryoid bodies at day 1 (Fig. 4A and B) and day 10 (Fig. 4C and D). The embryoid bodies from these hybrids were morphologically very similar to hESCs. When the embryoid bodies of hESC hybrids were further cultured in α-MEM supplemented with 10% FBS without feeder cells, the embryoid bodies spontaneously differentiated into several morphologically different cell types. Neural cell-like (Fig. 4E and F), mesenchymal cell-like (Fig. 4G and H), epithelium cell-like (Fig. 4I and J), and myocyte cell-like cells (Fig. 4K and L) that developed from hESC-hepatocyte hybrid cells were demonstrated.
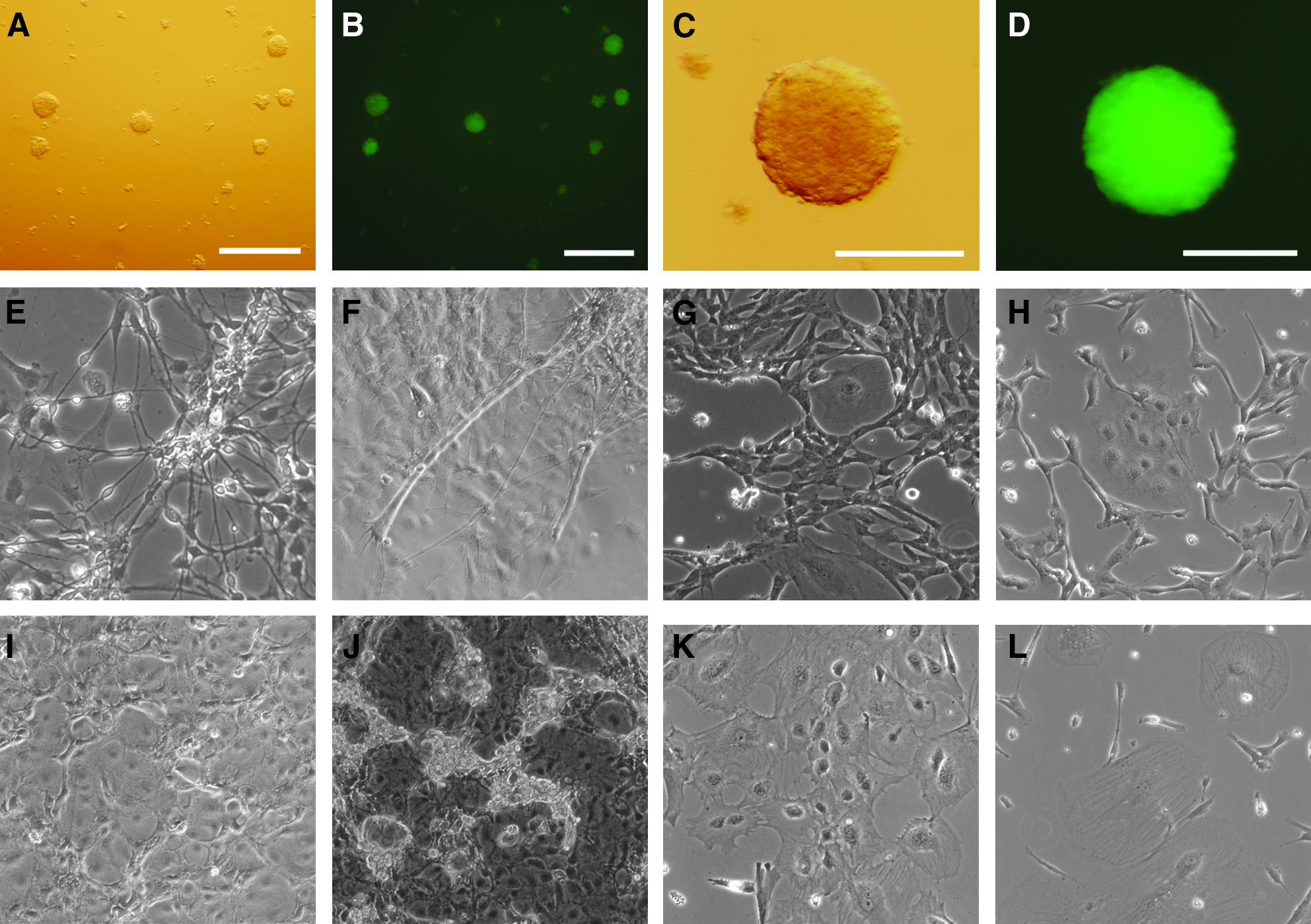
In vitro differentiation of hESC hybrids. Embryoid bodies of ENVY–hNHeps hybrid cells formed without feeder cells at day 1 (
Generation of hESC cybrids
The hESCs cultured on Matrigel-treated Thermanox plastic coverslips without feeder cells in condition medium were attached after 24 h (Fig. 5A and D). Enucleation efficiency of hESCs was evaluated by nuclear staining with Hoechst 33342 after centrifugation (Fig. 5 B and E, C and F). There were no significant differences in enucleation rates when centrifugation of the coverslips was performed in 10% Ficoll-400 at 20,000 (89.4%), 25,000 (90.0%), 30,000 (90%), or 35,000 (94%) RCF (Fig. 5G); however, a significantly lower enucleation rate was obtained from centrifugation at 25,000 RCF with 15.0% Ficoll-400 (68.0%) compared to 6.6% (98.5%), 8.0% (94.3%), or 10.0% (90.0%) (Fig. 5H) groups (p < 0.05). No significant differences in enucleation rates was detected between 6.6, 8.0, and 10.0% Ficoll-400 groups or 15.0 and 12.5% Ficoll-400 (78.7%) groups. Although the better enucleation was obtained from centrifugation medium containing 6.6% Ficoll-400, most cells detached from the coverslips after centrifugation; therefore, enucleation of hESCs was performed by centrifugation in 10% Ficoll-400 at 25,000 RCF for all the following experiments.

Generation of hESC cybrid cells by cell fusion of human fibroblasts and enucleated hESCs. hESCs, ENVY cells were cultured on Matrigel-treated coverslip without feeder cells before enucleation (
After fusion of enucleated ESC cytoplasts with human fibroblasts, ESC-like colonies formed. Detection of GFP in the cybrid colonies showed that some cybrid colonies were a mixed cell population with GFP-positive and GFP-negative cells at passage 0 (Fig. 5I and L, J and M). GFP-negative cell colonies at passage 1 were unable to proliferate in the same way as the GFP-positive colonies and ESC-like colonies (Fig. 5K and N).
Discussion
A number of research articles have been published on the reprogramming potential of mouse ESCs through cell fusion (Ambrosi et al., 2007; Do and Scholer, 2005; Silva et al., 2006). Collectively, fused hybrids of somatic cells and ESCs showed similarities to ESCs that are pluripotent, and they had the ability to differentiate into different cell types. It has also been reported that hESCs have the same potential as mouse ESCs to reprogram somatic cells into stem cell-like cells through cell fusion (Cowan et al., 2005; Yu et al., 2006). Hybrid cells can be easily produced by large-scale cell fusion; however, the resultant cells are usually a mixed population that includes hybrids and parent cells. A combination of double-antibiotics is commonly employed to culture the mixed cell population to remove both ESCs and somatic cells that have been transfected with different resistant genes. Such cells, however, are unlikely to be used for cell therapy due to the genetic modification that they have been subjected to. Therefore, it is important to generate pluripotent cells without genetic modification such as that achieved by cell fusion.
To avoid using genetic modification, the cell tracking dye SNARF-1 was evaluated in our experiments for hybrid selection by FACS. Cells labeled with SNARF-1 have a red–orange fluorescence that can easily be distinguished from that of cells loaded with green-fluorescent tracers. SNARF-1 is nontoxic to live cells, including embryos (Phillips et al., 1998), and has been successfully used for tracking of two cell populations with flow cytometry (Magg and Albert, 2007). The hESCs we used for fusion were electroporated with a vector that expressed GFP under the control of the human β-actin promoter and could also express high levels of GFP in all differentiated progeny (Costa et al., 2005).
Our results showed that the hybrids of fused hESCs and somatic cells loaded with GFP and SNARF-1 could be tracked and sorted by FACS. Carboxyfluorescein diacetate, succinimidyl ester (CFDA SE, V12883, Molecular Probes Invitrogen) combined with SNARF-1 to label hESCs and somatic cells was also able to track the fused hybrids by FACS (data not shown). Although the efficiency of cell fusion mediated by PEG/DMSO was low, a significant number of hybrid cells could be collected by FACS. These results indicate that the cell trackers can be used for generation of fused cell hybrids with the aid of FACS without gene transfection and drug selection. The ESC-like cells resulting from this method could potentially be applied to stem cell therapy.
Mouse neural stem cells (Do and Scholer, 2005; Matveeva et al., 2005), splenocytes (Matveeva et al., 1998; Vasilkova et al., 2007), thymocytes (Tada et al., 2003; Silva et al., 2006), cumulus cells (Do and Scholer, 2005), and fibroblasts (Matveeva et al., 2005; Silva et al., 2006) have been reprogrammed by ESCs. As for somatic stem cells (neural stem cells), fully differentiated somatic cells (cumulus cells) have been reprogrammed by ESCs through cell fusion (Do and Scholer, 2005). Human myeloid precursors differentiated from hESCs (Yu et al., 2006), pelvic bone cells (Cowan et al., 2005), and fibroblasts (Cowan et al., 2005; Yu et al., 2006) have also been reprogrammed by hESCs through cell fusion. We have now demonstrated that human hepatocytes are able to be reprogrammed by hESC hybrids to a certain extent, and these hybrid cells are similar to hESCs as previously characterized through expression of embryonic stem cell markers such as OCT4, TRA-1-60, TRA-1-81, SSEA-3, SSEA-4, and GCTM-2 (Thomson et al., 1998). However, expression of pluripotency markers was highest and most consistent at the edge of the colony. It has been reported that cells toward the edge of the colony are in intimate contact with the feeder cell layer, which produces stem cell maintenance factors and these factors drive stem cell maintenance (Hough et al., 2009). Peripheral staining of hybrid cells could have been resulted from lack of stem cell maintenance factors within the center of colonies.
SSEA-1, which is absent on hESCs and hESC-fibroblast hybrids and present in hepatocytes used in experiments (data not shown), was weakly detected in hESC–hepatocyte hybrids. However, alpha-fetoprotein, strongly expressed in hepatocytes, was turned off in hESC–hepatocyte hybrids. The results showed that SSEA-1 was still active in fused hybrids of hepatocytes and hESCs. Expression of SSEA-1 in hESCs indicates spontaneous differentiation (Henderson et al., 2002); however, in hESC–hepatocyte hybrids it may indicate that the hybrids were not fully reprogrammed back to embryonic stem cell status as indicated by the weak expression of SSEA-1 from hepatocytes. These results demonstrate that somatic cells can be reprogrammed into stem cell-like cells by hESCs; however, reprogrammed cell hybrids are not identical to ESCs. The genes implicated in pluripotency and chromatin function are expressed; however, large numbers of somatic genes are silenced in hybrids and ESCs (Ambrosi et al., 2007). Although it is possible to generate human pluripotent hybrid cells by cell fusion, the pluripotency of such a fused hybrid cell may derive entirely from the pluripotent ESC genome because the somatic genome is controlled by the pluripotent cell genome (Do et al., 2007).
Karyotype analysis of fused hybrids showed variable chromosome numbers, especially when the selection antibiotics were removed from the culture medium (Serov et al., 2003). Aneuploid chromosomes were frequently observed in most reprogrammed hybrid cells, although most of them were near-tetraploid (Ambrosi et al., 2007; Matveeva et al., 1998; Serov et al., 2003; Vasilkova et al., 2007; Yu et al., 2006). The phenomenon of aneuploidy not only occurred in hybrid cells fused in vitro, but also in hybrid cells spontaneously fused in vivo (Terada et al., 2002; Wang et al., 2003). In our study, a large number of aneuploid hybrid cells were produced by cell fusion without drug selection, which was similar to that observed for mouse ESC hybrids (Mittmann et al., 2002). It appears that chromosome numbers of fused ESC hybrids derived from male and female cells are much more close to euploid (Matveeva et al., 1998; Tada et al., 2001, 2003). Except for fused ESC hybrids, it was found that female hESCs were more difficult to maintain their undifferentiated state in the culture dish comparing with male hESCs (data not included). Additionally, human hepatocytes are naturally polyploidized during normal development (Kudryavtsev et al., 1993). This is a protective mechanism associated with many physiologic and pathologic processes directed against the harmful effects of oxidative stress that occurs via a controlled process throughout growth and aging where binucleation is important (Lu et al., 2007). That probably is the reason why hepatocyte hybrids exhibit large percentage of aneuploidy.
Fused hybrid cell is pluripotential because the chromosomes of mouse ESC are dominant in the fused cell hybrid, in which only a few of chromosomes are from the somatic cell (Vasilkova et al., 2007). In our experiments, the fused cell hybrids that have near-diploid and near-tetraploid chromosomes may have been derived from where hESC chromatin was dominant and contained only a few of chromosomes from hepatocytes. Further investigation is needed to clarify which chromosomes are dominant in hESC hybrids.
Reprogrammed ESC hybrids have the potential to differentiate into all three germ layers in mice (Ambrosi et al., 2007; Do and Scholer, 2005) and humans (Cowan et al., 2005; Yu et al., 2006), and can also contribute to chimeras despite near-tetraploid chromosomes (Kruglova et al., 2008; Matveeva et al., 1998; Tada et al., 2001). Consistent with these findings, embryoid bodies and different types of differentiated cells from the fused cell hybrids, especially from hESC–hepatocyte hybrids, were produced in this study.
Enucleation of hESCs is a critical step for generating hESC cybrid cells. Due to the characteristics of hESCs, bulk enucleation is the only way to enucleate hESCs. Thus, it is important to culture hESCs in monolayer without feeder cells. It has been found that culture of hESCs was very efficient on Matrigel-treated coverslips without feeder cells to form monolayer culture in 24 h, and were able to be enucleated afterwards in this study. However, hESCs were unable to be enucleated when compact multilayer cells formed after further culture. Because cell enucleation was reported in 1967 (Carter, 1967), enucleation of mammalian cells has been widely used for a variety of studies (Poste, 1973). The cytoplast maintains function for a short time after enucleation (Degaetano and Schindler, 1987; England et al., 1978; Goldman et al., 1973; Shay et al., 1974), and the anucleate cells have the potential to reconstruct into cell hybrids (Ladda and Estensen, 1970; Poste and Reeve, 1972; Veomett et al., 1974) and cybrids (Shay, 1987; Shay and Clark, 1980; Strelchenko et al., 2006). We found that high efficient enucleation (almost 100%) could be achieved in low concentrations of Ficoll-400, but the resultant cytoplasts were too small to fuse with somatic cells (data not shown). Increasing concentration of Ficoll-400 to 10%, about 91% of cells were enucleated and this concentration was used for in our experiments. Consequently, a mixed cell population containing fused cell hybrids, cybrids, and nonfused hESCs and somatic cells was generated. Hence, purification of cytoplasts needs to be further improved.
To generate hESC cybrids, enucleated hESCs were fused with fibroblast cells in our studies, and cell fusion was performed by PEG/DMSO. The hESC cybrids were generated by the same way as the hybrid generation despite low fusion rate. Similar results were also obtained by fusion of enucleated hESCs with fibroblast cells on coverslips after bulk enucleation centrifugation without dissociation of enucleated hESCs from coverslip (data not included).
Only one report has shown that somatic cells were successful reprogrammed by enucleated hESCs through cell fusion, but the fused hESC cybrids were a mixed cell population containing diploid 46, XY, near-diploid 46, XXY, and near-tetraploid 92, XXYY cells (Strelchenko et al., 2006). Similar experiments were unsuccessful in mice (Do and Scholer, 2004). Consistent with the results from mouse ESC cybrids, we found that fused hESC cybrids of hepatocytes or fibroblasts were unable to self-renewal in the way that hESC colonies. Only the cybrids mixed with nucleated hESCs that presented partial GFP-positive cells were able to form colonies that could be passaged. Although pure hESC cybrids, which were totally GFP-negative colonies, could be generated after cell fusion, the GFP-negative colonies were unable to form colonies after passage. It has been suggested that nuclear components of oocytes may contain some cell reprogramming factors (Chang et al., 2004; Gao et al., 2002) and such reprogramming factors were incorporated into pronuclei after fertilization (Polanski et al., 2005). Thus, removal of the condensed chromosomes of the zygote during mitosis successfully generates cloned animals, but removal of the pronucleus of the zygote fails (Egli et al., 2007; Howlett et al., 1987; Illmensee and Hoppe, 1981; McGrath and Solter, 1983; Tsunoda et al., 1987; Wakayama et al., 2000). We presume that the important reprogramming factors in hESC are likely incorporated into the nucleus, and that the relevant reprogramming factors are removed when hESCs are enucleated. Thus, the enucleated hESCs have limited potential to reprogram somatic cells evidenced by limited self-renewal capacity of mixed-cell population with hESCs, however not in purified cybrid cells. Although hESC cybrid cells were still able to form small colony after fusion with somatic cells, self-renewal capacity did not exist, indicating that some reprogramming factors remained in the cytoplast after enucleation; however, they were not able to support proliferation. It will be of interest to investigate the reprogramming potential of hESC cytoplasts if suitable protocols are available for enucleating hESCs during mitosis.
Conclusions
The hESCs, but not hESC, cytoplasts have the potential to reprogram human fibroblasts and hepatocytes into ESC-like cells through cell fusion. This indicates that the important reprogramming factors are associated with karyoplast and are not present in cytoplast. The reprogramming of hybrid cells does not fully recapitulate the ESC state, and frequently resulted in aneuploid chromosomes, even though the hESC hybrids were able to differentiate into embryoid bodies and some somatic cell types in vitro.
Footnotes
Acknowledgments
The authors thank Andrew Fryga for expert assistance with flow cytometry analysis and sorting, Dr. Jinhua Li for assistance with photography using the Olympus Fluoview 1000 confocal microscope and FV10-ASW software. This research was supported by an innovative grant funded by the Juvenile Diabetes Research Foundation (File No: 5-2006-203).
Author Disclose Statement
The authors declare that no conflicting financial intersts exist.