
Abstract
Abstract
Deriving histocompatible embryonic stem (ES) cells by somatic cell nuclear transfer (SCNT) and parthenogenetic activation (PA) requires fresh oocytes, which prevents their applications in humans. Here, we evaluated the efficiency of deriving ES cells from mature metaphase II (MII) and immature metaphase I (MI) vitrified oocytes, by PA or SCNT, in a mouse model. We successfully generated ES cell lines from PA (MII and MI) and SCNT (MII and MI) blastocysts. These cell lines expressed genes and antigens characteristic of pluripotent ES cells and produced full-term pups upon tetraploid embryo complementation. This study established an animal model for efficient generation of patient-specific ES cell lines using cryopreserved oocytes. This is a major step forward in the application of therapeutic cloning and parthenogenetic technology in human regenerative medicine and will serve as an important alternative to the iPS cell technology in countries/regions where these technologies are permitted.
Introduction
The use of excess oocytes that already are frozen and stored in human fertility clinics and immature oocytes removed in preparation for IVF may be the answer. It has been more than 20 years since the birth of the first human child conceived through the fertilization of a frozen and thawed metaphase II (MII) oocyte (Chen, 1986), and only recently, cryopreservation of human oocytes has become sufficiently efficient for clinical applications. Although 60–100 frozen and thawed oocytes were used to achieve one pregnancy in the late 1990s (Porcu et al., 1999; Tucker et al., 1998), only 5–13 are now needed to achieve an implantation (Antinori et al., 2007; Barritt et al., 2007; Kuwayama et al., 2005). Because this efficiency (∼13 oocytes/implantation) is comparable to that of fresh oocytes (∼10 oocytes/implantation) (Antinori et al., 2007), oocyte cryopreservation is finally being incorporated into human IVF. It also serves as an option to preserve female gametes before a patient undergoes invasive medical treatments, such as surgical removal of the ovaries, radiotherapy, and/or chemotherapy. With recent improvements in assisted reproductive technologies, fewer oocytes are needed to achieve patients' goals of reproduction; thus, substantial numbers of unused frozen eggs are stored in fertility clinics worldwide. These eggs could become available for donation and used to generate ES cell lines.
When performing controlled ovarian stimulation, a portion (15–20%) of human oocytes remains meiotically immature (first metaphase or MI phase) (Cha and Chian, 1998; Smitz and Cortvrindt, 2004). These immature oocytes can be matured in vitro to the MII phase after 20–36 h of culture. Because the additional culture time causes asynchrony, most IVF centers do not practice oocyte in vitro maturation or make further attempts at fertilizing these immature oocytes. As a result, most of the immature oocytes are routinely discarded in human IVF cycles. These immature oocytes can also be donated for research with the patients' consents and proper IRB approvals, making them an additional source for cryopreserved human oocytes for ES cell derivation.
With minimal medical, ethical, and logistical concerns, cryopreserved mature and immature oocytes may become the solution to the shortage of human oocytes for human stem cell research. However, it has not been demonstrated that frozen and thawed oocytes contain full nuclear reprogramming activity and will support ES cell derivation. Before testing cryopreserved human oocytes, it is important to examine the feasibility in an animal model. In this study, we performed a proof-of-principle experiment in mice to test the validity and efficiency of the SCNT-ES and PA-ES cell derivation from vitrified mature and immature oocytes.
Materials and Methods
Chemicals and culture media
Unless otherwise indicated, all chemicals were purchased from Sigma Chemical Co. (St. Louis, MO). All media were prepared fresh and filter-sterilized through a 0.22-μm filter (Acrodisc; Pall Gelman Laboratory, Ann Arbor, MI).
Recovery of metaphase II and I oocytes
The B6D2F1 (C57BL/6 X DBA/2) mice used were from Charles River Laboratories (Wilmington, MA, USA) and The Jackson Laboratory (Bar Harbor, ME, USA). All experimental animal procedures were approved by the Institutional Animal Care and Use Committees of the University of Connecticut.
We collected in vivo matured MII-stage oocytes from B6D2F1 female mice subjected to the following hormone priming protocol: superovulation was induced with 7.5 IU of equine chorionic gonadotrophin (eCG), followed 48 h later with 7.5 IU of human chorionic gonadotrophin (hCG). Oocytes at the MI stage were collected 6–7 h after the hCG treatment by puncturing mature follicles and freeing them of cumulus cells by 2 min exposure to 0.1 mg/mL of hyaluronidase at 37°C and pipetting. Oocytes at the MII stage were harvested 13–14 h after the hCG treatment and freed of cumulus cells by 2-min exposure to 0.1 mg/mL of hyaluronidase at 37°C and gentle pipetting.
Vitrification of oocytes
The basal medium used for oocyte cryopreservation was HEPES-buffered human tubal fluid (HTF) supplemented with 20% (v/v) fetal bovine serum (FBS; Hyclone, Logan, UT, USA). The denuded oocytes were vitrified by the minimum volume cooling method, essentially as described by Kuwayama et al. (2005). Briefly, the oocytes were equilibrated in equilibration medium [basal medium with 7.5% (v/v) ethylene glycol and 7.5% (v/v) dimethylsulphoxide (DMSO)] at room temperature for 5 min. The oocytes were then transferred into the vitrification medium [basal medium with 15% (v/v) ethylene glycol, 15% (v/v) DMSO, and 0.5 mol/L sucrose] at room temperature (RT) for 45–60 sec. The cryoprotectant-treated oocytes were placed onto a fine polypropylene strip (Cryotop®, Kitazato BioPharma Co., Fuji, Shizuoka, Japan). The polypropylene strip carrying the oocytes was then submerged into liquid nitrogen and was ready for storage.
Thawing procedures
The polypropylene strip with vitrified oocytes was immersed directly into 5.0 mL of thawing solution [HEPES-buffered HTF with 20% (v/v) FBS and 1.0 mol/L sucrose] at 37°C for 1 min. Oocytes were then picked up and transferred into 1.0 mL of the dilution solution [HEPES-buffered HTF with 20% (v/v) FBS and 0.5 mol/L sucrose] for 3 min at RT. The oocytes were subsequently washed in 1.0 mL washing solution [HEPES-buffered HTF with 20% (v/v) FBS] for 10 min at RT. Finally, the oocytes were incubated in KSOM + AA medium (Specialty Media, Phillipsburg, NJ, USA) before oocyte activation or nuclear transfer.
In vitro culture
When cryopreserved MI oocytes were subjected to nuclear transfer, the frozen/thawed MI oocytes were directly culture in KSOM + AA medium for 6 h to extrude the first polar body and became matured MII oocytes. Only oocytes at MII phase were used as recipients for nuclear transfer. For parthenogenetic activation, frozen MI oocytes were thawed, and subsequently cultured in KSOM + AA medium containing 5 μg/mL cytochalasin D (CD) for 3 h, then further cultured without CD for 5 h in vitro prior to activation.
Somatic cell nuclear transfer (SCNT), parthenogenetic activation (PA), and embryo culture
SCNT was performed by the fusion method (Ogura et al., 2000). For enucleation, groups of 20–30 oocytes were transferred to HEPES-buffered CZB medium containing 5 μg/mL cytochalasin B (CB). The spindle chromosome complex (SCC) was removed using a pipette with an inner diameter of 8–10 μm assisted by piezo-drill pulses. Nuclei from tail-tip fibroblast cells of B6D2F1 mice were transferred by electrofusion to produce cloned blastocysts. A fibroblast cell with an approximate diameter of 15–20 μm was selected and inserted into the perivitelline space of an enucleated oocyte. Following transfer, the cell-cytoplast complexes were induced by a DC pulse of 2500 V/cm for 10 μsec by an Electrocell Manipulator 200 (BTX, San Diego, CA, USA) in 280 mM mannitol containing 0.1 mM MgSO4, 0.1 mg/mL polyvinyl alcohol, and 3 mg/mL bovine serum albumin. An hour later, fusion was confirmed by microscopic analysis.
Fused pairs were activated in calcium-free CZB medium containing 10 mM strontium, 5 μg/mL cytochalasin B, and 10 nM trichostatin A (TSA) for 6 h. Following activation, cloned embryos were cultured in KSOM + AA medium containing 10 nM TSA for 4 more hours, then further cultured in KSOM + AA medium for 4 days at 37°C in a humidified atmosphere of 5% CO2, 5% O2, and 90% N2. For parthenogenetic activation, frozen/thawed oocytes at the MI phase were in vitro cultured 3 h with CD and 5 h without CD. These oocytes were activated in KSOM + AA medium containing 10 mM strontium for another 6 h. The oocytes at the MII phase were activated in KSOM + AA medium containing 10 mM strontium and 5 μg/mL cytochalasin B for 6 h. After activation, oocytes were cultured in KSOM + AA medium for 4 days, as described for cloned embryos, above.
Establishment ES cell lines
When embryos reached the blastocyst stage after 4 days of culture, they were used to establish ES cell lines, as described by Wakayama et al. (2001). Briefly, cloned and parthenogenetically activated blastocysts were placed into a 96-well plate containing inactive primary mouse embryonic fibroblast (pMEF) feeder cells and cultured for about 10 days. Proliferating outgrowths were dissociated using trypsin (Invitrogen, Carlsbad, CA, USA) and replated on pMEF feeder cells until stable cell lines grew out. Dulbecco's Modified Eagle Medium (DMEM) for ES cell culture was purchased from Specialty Media and was supplemented with 15% knockout serum replacement (Invitrogen) and 1000 U/mL leukemia inhibitory factor (LIF) (Invitrogen), plus 1% penicillin–streptomycin (Invitrogen), 1% L-glutamine (Specialty Media), 1% nonessential animal acids (Specialty Media), 1% nucleosides for ES cells (Specialty Media), and 1% 2-mercaptoethanol (Specialty Media). Medium was changed every day and cells were split every 2–3 days.
In vitro characterization of ES cells
Pluripotency of the established ES cell lines was determined by alkaline phosphatase staining, immunohistochemistry, and RT-PCR of ES cell markers of Oct4, Nanog, and Sox2. Immunohistochemistry was performed using the following antibodies: anti-Oct4 (MAB4401, Millipore, Bedform, MA, USA), anti-Nanog (AB9220, Millipore), anti-Sox2 (MAB4343, Millipore), and antistage-specific embryonic antigen 1 (SSEA1, MAB4301, Millipore). Karyotype assay was performed to determine the chromosome normality of ES cell lines.
Blastocyst injection
CD-1 strain mice were used to collect diploid or tetraploid blastocysts (94–98h post-hCG). Tetraploid embryos were generated by electrofusion of two-cell embryos collected from fertilized eggs, as described previously (Amano et al., 2009). For blastocyst injections, groups of 10–20 blastocysts were transferred to a drop of HEPES-buffered CZB medium under mineral oil, and 10 to 15 ES cells were injected into blastocysts using a flat tip pipette with an inner diameter of 14–16 μm assisted by piezo-drill pulses. After injection of the entire group, blastocysts were returned to KSOM + AA medium at 37°C until transfer to recipient females. Embryos were transferred into the uteri of day 2.5 pseudopregnant CD-1 females mated with vasectomized males. The full-term pups were obtained from recipient mothers at day 18.5 by Caesarean section.
Statistical analysis
Statistical analysis of the data on development of PA and SCNT embryos was carried out using the Generalized Linear Model procedures (PROC GENMOD in SAS).
Results
Survival of cryopreserved MI and MII oocytes
We obtained an excellent survival rate (97.8%; 739/756; Table 1) for mouse MII oocytes (Fig. 1A) after freezing by vitrification and thawing (Fig. 1B), as judged by oocyte membrane integrity. Only viable oocytes with healthy, smooth, and intact membranes were selected for further experiments (Fig. 1C). We also tested the viability of MI oocytes after freezing/thawing procedures; the survival rate was 93.4% (809/866). After 6 h of in vitro maturation, 93.3% (754/809) of oocytes extruded the first polar body and became mature MII oocytes (Table 1).
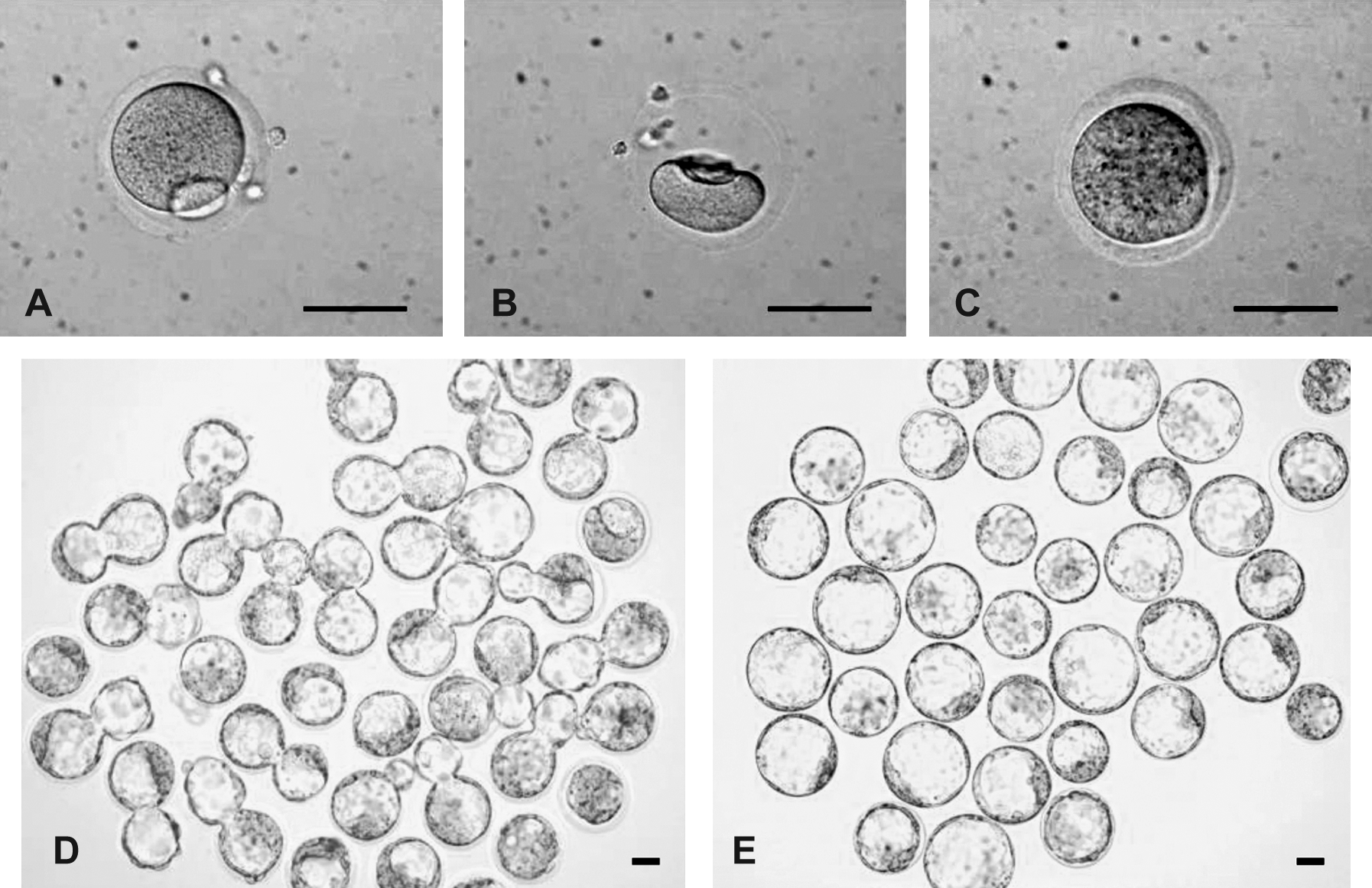
Development of SCNT and parthenogenetic embryos from cryopreserved eggs. (
Nuclear reprogramming by cryopreserved mature (MII) oocytes
To determine if the cryopreserved oocytes retained the capability of nuclear reprogramming, we first compared the efficiencies of preimplantation development by SCNT using fresh (control) and cryopreserved MII oocytes (Table 2). Comparisons were also made to diploid parthenogenetic B6D2F1 embryos derived from fresh and cryopreserved oocytes. As expected, higher rates of enucleation survival (99.6 vs. 91.6%) and somatic cell-cytoplast fusion (88.5 vs. 71.3%) were observed for fresh oocytes than for cryopreserved oocytes in SCNT trials. However, cloned embryos from both fresh and cryopreserved oocytes were able to develop to the blastocyst stage (Table 2, Fig. 1D), albeit there was a 23.8% (56.3 vs. 80.1%) decrease in this ability for cryopreserved oocytes versus fresh oocytes. In parthenogenetic derivation, 97.3% (181/186) of the fresh oocytes developed to blastocysts. The difference between achievements of blastocyst development was only 11.8% (85.5 vs. 97.3%) in the PA group when cryopreserved oocytes and fresh oocytes were compared (Fig. 1E). The decreased developmental potential was likely a result of cryo-injuries during the freezing and thawing processes (Table 2).
Different superscripts within columns indicate a significant difference between treatment groups (p < 0.05).
PA, parthenogenetic activation; SCNT, somatic cell nuclear transfer; cleaved: two- to four-cell stage embryos; M + B, morula and blastocyst stage embryos.
Nuclear reprogramming by cryopreserved immature (MI) oocytes
We further examined whether immature MI oocytes could be used as recipients for SCNT after cryopreservation and culture in vitro. As shown in Table 3, cryopreserved MI oocytes after thawing and culture in vitro to the MII phase had a similar ability to vitrified MII oocytes (Table 2) to support the development of parthenogenetic (206/244 or 84.4% for MI vs. 247/289 or 85.5% for MII) and SCNT (101/198 or 51.0% for MI vs. 108/192 or 56.3% for MII) embryos to the blastocyst stage.
Different superscripts within columns indicate a significant difference between treatment groups (p < 0.05). For MII oocyte controls, please refer to Table 2.
Frozen MI oocytes were thawed, and subsequently cultured with cytochalasin D (CD) for 3 h, then further cultured without CD for 5 h in vitro prior to activation.
Frozen MI oocytes were thawed, and subsequently cultured for 6 h in vitro. Only oocytes at MII phase were used as recipients for SCNT.
PA, parthenogenetic activation; SCNT, somatic cell nuclear transfer; cleaved: two- to four-cell stage embryos; M + B, morula and blastocyst stage embryos.
Derivation of histocompatible PA-ES and SCNT-ES cell lines from cryopreserved MII and MI oocytes
We next determined whether ES cell lines could be obtained from embryos derived from cryopreserved oocytes. We transferred 123 MII-derived and 187 MI-derived diploid parthenogenetic blastocysts into an ES cell culture system; 87 (70.7%; 87/123) and 103 (55.1%; 103/187) of the blastocysts attached to the fetal fibroblast feeder layer with outgrowths of inner cell mass (ICM), and 83 (67.5%; 83/123) and 97 (51.9%; 97/187) of the respective outgrowths gave rise to ES cell lines that grew in colonies (Table 4). For SCNT embryos, 25 (24.8%; 25/101) and 34 (59.6%; 34/57) of SCNT-ES cell lines were established from cryopreserved MII and MI oocytes, respectively (Tables 5 and 6).
PA-ES
PA, parthenogenetic activation; ICM, inner cell mass; ET, embryo transfer; MII—PA, frozen oocytes at MII phase were subject to activation immediately after thaw; MI—PA, frozen oocytes at MI phase were thawed, and cultured with CD for 3 h, then further cultured without CD for 5h in vitro prior to activation.
SCNT-ES
PA, parthenogenetic activation; SCNT, somatic cell nuclear transfer; ICM, inner cell mass; ET, embryo transfer.
MI oocytes were frozen, thawed, and then culture for 6 h in vitro. Only oocytes at MII phase were used as recipients for SCNT.
MII-SCNT-ESC, ESC lines derived from fresh (control) MII oocytes by nuclear transfer; F/T-MII-SCNT-ESC, ESC lines derived from frozen and thawed MII oocytes by nuclear transfer; F/T- MII-PA-ESC, ESC lines derived from frozen and thawed MII oocytes after parthenogenetic activation (PA); F/T-MI-SCNT-ESC, ESC lines established from SCNT embryos, which were derived from frozen/thawed MI oocytes after 6 h culture in vitro to MII phase prior to SCNT; F/T- MI-PA-ESC, ESC lines derived from frozen and thawed MI oocytes after PA.
Three ES cell lines from each of the following groups—PA frozen MII, SCNT frozen MII, PA frozen MI, and SCNT frozen MI—were selected at random for examination of pluripotency. The mRNA for Oct4, Nanog, and Sox2, were expressed in all of these ES cells (Fig. 2B) but not in skin fibroblasts (data not shown). These cell lines were also immunoreactive to antibodies specific for Oct4, Nanog, Sox2, and the stage-specific embryonic antigen-1 (SSEA1) (Fig. 2A). Karyotyping revealed the average of 71.8 ± 5.8%, 76.6 ± 4.3%, 72.8 ± 6.2%, and 80.3 ± 5.2% of normal 40XX or 40XY karyotype (Fig. 2C) in cryopreserved MI-PA-ES, MI-SCNT-ES, MII-PA-ES, and MII-SCNT-ES cell lines, respectively.
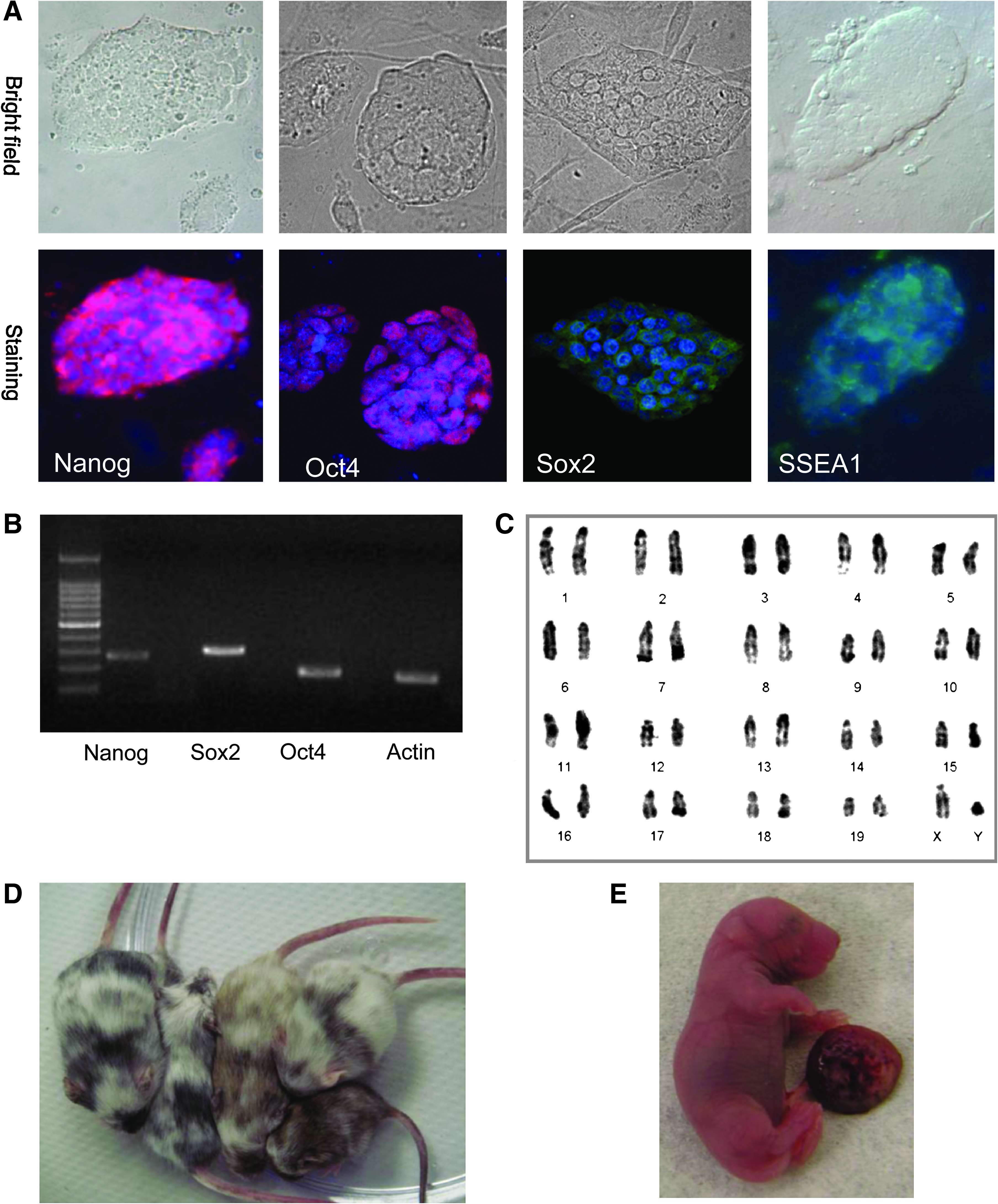
In vitro and in vivo characterizations of ES cell lines derived from cryopreserved eggs. Three SCNT-ES cell lines were characterized by RT-PCR, immunostaining, and karyotyping. (
The in vivo developmental potential of the PA-ES cell lines was determined by the percentage of chimeras from diploid blastocyst injections; the SCNT-ES cell lines were verified through tetraploid embryo complementation. For PA-ES cells, we generated 34 (29.3%; 34/116) and 50 (38.5%; 50/130) chimeric mice by injecting MII-PA-ES and MI-PA-ES cell lines into CD1 (white coat) diploid blastocysts. A high level of black coat color chimerism was observed in the PA-ES cells from BDF-1 mice (black coat; Fig. 2D). This result was comparable to that for parthenogenetic ES cell lines derived from fresh oocytes, as reported in a prior study (Kim et al., 2007a). For SCNT-ES cell lines, we produced seven (2.0%; 7/346) and 2 (0.9%; 2/230) full-term pups, composed only of the injected donor MII and MI-derived SCNT-ES cells, by tetraploid embryo complementation (Fig. 2E). These findings suggest that the cell lines derived from cryopreserved eggs can develop into all lineages of the embryonic germ layers and are thus as pluripotent as conventional ES cells.
Discussion
Because of their large size and complex subcellular structures, oocytes incur more detrimental damages from cryopreservation than much smaller cells, such as the sperm or somatic cells. These effects can significantly compromise the oocyte's fertility (Gardner et al., 2007). In mouse studies, cryoprotectants have been shown to cause a large transient increase in intracellular calcium(Larman et al., 2006), a decrease in maturation promoting factor (MPF) activity, and, subsequently, spontaneous activation or degeneration of the oocyte (Larman et al., 2007; Shaw and Trounson, 1989; Van der Elst et al., 1988). By using a modified Cryotop® vitrification protocol, we were able to cryopreserve oocytes with only minor damage to their quality (Tables 2 and 3), as judged by the rates of post-thawing survival and, more importantly, by the rates of blastocyst formation after SCNT and PA. For instance, only a 10% decrease was caused by cryo-injury in the MII-PA group (fresh-MII vs. cryopreserved-MII 97.3 vs. 85.5%). These results are higher than those reported in other studies following parthenogenetic activation (58–62% of blastocyst rate) (Endoh et al., 2007) or fertilization (65% of blastocyst rate) (Eroglu et al., 2009) in the mouse or in humans for cryosurvival (20–80%), and fertilization (30–60%) (Fabbri et al., 2000). Although recently, Antinori et al. (2007) reported high human oocyte cryosurvival (99.4%) and fertilization rates (92.9%), the pregnancy and implantation rates per embryo were low (32.5 and 13.2%, respectively). More importantly, our vitrification process does not greatly diminish the nuclear reprogramming ability of the oocyte. The potential of SCNT blastocyst formation from cryopreserved oocytes was only reduced to ∼70% (fresh-MII vs. cryopreserved-MII: 80.1 vs. 56.3%) of that of the control oocytes. Embryos from cryopreserved oocytes appeared to have recovered completely by the blastocyst stage because the efficiency of SCNT-ES cell derivation from cryopreserved oocytes is equivalent to that from fresh oocytes (Kishigami et al., 2006; Wakayama et al., 2001). These results demonstrated that frozen MII oocytes have the potential to be good resources for SCNT and ES cell research and biomedical applications.
In addition to excess frozen matured oocytes, immature oocytes are the next most readily available human oocyte source, making them candidates for use in stem cell research and therapeutic cloning. Cryopreservation can preserve these valuable materials (Antinori et al., 2007; Barritt et al., 2007; Borini et al., 2006; Kuwayama et al., 2005; Oktay et al., 2006). In our mouse model, we examined whether ES cell lines could be generated using embryos derived from cryopreserved MI immature oocytes post-thawing and culture in vitro. To our surprise, 84.4% (206/244) and 51.0% (101/198) of the resulting parthenogenetic and SCNT embryos, respectively, developed to the blastocyst stage (Table 3), the data shown was no different with that which was derived from fresh MII or MI oocytes (MII-parthenogenetic: 74%, Kim et al., 2007a; MI-parthenogenetic: 56%, Kim et al., 2007a; MII-SCNT: 40–60%, Hochedlinger and Jaenisch, 2003; Kishigami et al., 2006). Moreover, the ES cell derivation rates were as high as 51.9% (97/187) and 59.6% (34/57) for PA and SCNT embryos, respectively (Tables 4 and 5); the results were also comparable with that which was derived from fresh oocytes (MII-PA-ESC: 65% and MI-PA-ESC: 37%, Kim et al., 2007a; MII-SCNT: 33%, Kishigami et al., 2006). To our knowledge, this is first report in which frozen mature MII and immature MI oocytes were used for ESC generation. Our data suggest that the developmental potential and the reprogramability of vitrified MI oocytes by blastocyst or ESC lines generation were comparable to those from fresh MI and even fresh MII oocyte in both nuclear transfer (Kishigami et al., 2006) or parthenogenetic activation (Kim et al., 2007a). As summarized in Table 6, although 11 and 2 cryopreserved MII oocytes were needed to produce an SCNT or parthenogenetic ESC line, only 5 and 2 cryopreserved MI oocytes were necessary for the same outcome. All cell lines, either from cryopreserved MII or MI oocytes, were fully competent upon in vitro and in vivo evaluation for development and differentiation potentials as those reported for fresh MII oocytes (Eggan et al., 2004; Kim et al., 2007a; Wakayama et al., 2001).
To date, several different approaches have been developed for the purpose of nuclear reprogramming: (1) SCNT into unfertilized oocytes or mitotic zygotes (Egli et al., 2007; Wakayama et al., 1998; Wilmut et al., 1997)’ (2) fusion of embryonic stem cell to somatic cells (Cowan et al., 2005; Tada et al., 2001); (3) transfection of extracts of eggs or pluripotent cells to somatic cells (Hansis et al., 2004; Kikyo et al., 2000; Tamada et al., 2006; Taranger et al., 2005); and (4) induction of pluripotent cells (iPS) by exogenous expression of pluripotency factors (Oct3/4, Sox2, Klf4, and c-Myc) (Okita et al., 2007; Takahashi and Yamanaka, 2006; Wernig et al., 2007). All of these approaches, however, are associated with ethical, safety (foreign gene transfer and use of virus), or efficacy (the degree of reprogramming) concerns. Among them, SCNT using unfertilized oocytes has been shown in proof-of-principle mouse studies to be of therapeutic value (Rideout et al., 2002; Tabar et al., 2008). Moreover, ES cells derived from SCNT blastocysts have been demonstrated to be transcriptionally and functionally indistinguishable from those derived from fertilized blastocysts (Brambrink et al., 2006). Although the generation of hESC from SCNT embryo has not been successful, a parthenogenetic ESCs from human embryo has been found to be able to generate histocompatible tissues for transplantation. Due to genomic imprinting disruptions, PA-ES cells from fresh MII and MI oocytes have been reported to have no germline transmission ability (Kim et al., 2007a). This, however, does not diminish the value of PA-ES cells as a potential alternative for autologous tissue generation. Indeed, PA-ES cells have been shown to be nonhomozygous and do possess distinct signature of genetic recombination (Kim et al., 2007b) and stable and functional hematopoietic reconstitution (Eckardt et al., 2007). Together, our results and those of others demonstrated that parthenogenetic or SCNT ESC lines are important alternatives to the highly researched iPS technology for which the use of viral transfection still serves as a major road block for its clinical application.
In summary, our results demonstrated that somatic nuclei could be reprogrammed by vitrified oocytes at either the MII or MI phase after being thawed and cultured in vitro. Embryos so produced could be used to derive pluripotent ES cell lines. These findings present the possibility of using demonstrated human oocytes for generating patient-specific ES cell lines. By overcoming the shortage of fresh human oocytes, SCNT can become one of the most promising therapeutic approaches in regenerative medicine.
Footnotes
Acknowledgments
We appreciate the technical assistance provide by Tuz-An Lin. This research was supported by funds from USDA-ARS to X.C.T. and X.Y. and to L.Y.S. from National Taiwan University and National Science Council (NSC 96-2321-B-002-033).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
The first three authors contributed equally to this work.