
Abstract
Abstract
Despite recent cell-engineering advances, treatment and repair of cartilage remains challenging. Although stem cell transplantation therapy using mesenchymal stem cells (MSCs) is considered a prominent strategy, the major problem of limited proliferative capacity of autologous cells has been unsolved. Recently, an induced pluripotent stem (iPS) cell line was suggested as an alternative way to cure various human diseases due to their potential proliferating infinitely while possessing the capacity to form all types of cells. However, the method to induce lineage-restricted differentiation has not been well examined or established. Here, we suggest a simple method to induce mesenchymal progenitors possessing chondrogenic property from mouse iPS cells. The MSC-like cells produced in our study expressed some MSC markers, and could also differentiate to osteoblast and adipocyte. The present study demonstrates the property of iPS cells as an alternative candidate for treatment of articular disorders, and suggests an effective approach for preparing chondrocyte from iPS cells.
Introduction
Recently, novel pluripotent stem cell lines, termed induced pluripotent stem (iPS) cells, were generated from mouse skin fibroblasts by introducing pluripotent-related transcription factors such as Oct3/4, Sox-2, and Klf-4 (Okita et al., 2007; Takahashi and Yamanaka, 2006). They were also successfully generated from human skin fibroblaststs (Takahashi et al., 2007; Yu et al., 2007). The iPS cells represent promising cell sources for transplantation because of their unlimited self-renewal property and ability to differentiate into various somatic cell lineages. The medical use of the iPS cells can evade potential ethical problems, which are inevitable in application of embryonic stem (ES) cells, for example, the use of embryos that possess developmental potential to term. Furthermore, the iPS cells could be made directly from patients without harming the cell donor. Pluripotent stem cells such as ES cells and iPS cells differentiate into cell types of all three embryonic germ layers when cultivated as cell aggregates called an embryoid body (EB). Many reports have shown that the system recapitulates cellular developmental processes and gene expression patterns of early embryogenesis.
Chondrocyte differentiation from ES cells via EB formation have already been reported by many groups; encapsulation of the EBs in scaffold have been suggested as an effective way for obtaining chondrocytes from the pluripotent cells (Fecek et al., 2008; Hwang et al., 2006; Jukes et al., 2008). On the other hand, differentiation of ES cells to EBs results in heterogeneous populations of differentiated cells. Heterogeneous status of the transplant may lead to inferior tissue function and organization of engineered tissues (Hwang et al., 2008; Koay et al., 2007). Cell sorting using flow cytometry is one of the effective ways for selection of specific cell types. Nakayama et al. (2003) sorted mesenchymal progenitors from EBs induced from mouse ES cells using a specific antibody to platelet-derived growth factor receptor alpha (PDGFRα), which is expressed in most of the mesodermal cells including the axial skeleton during early embryogenesis, as a marker and demonstrated that the PDGFRα + cells possess high chondro-differentiation capacity. However, the procedure is very troublesome, and it may make it difficult to attain some critical factors for clinical uses such as absolute sterility, cleanliness, reliability, and protection of the sort operator from aerosolized particles (Ibrahim and van den Engh, 2003). Moreover, the sorting process entails an important cell loss and damages. Other cell-sorting techniques such as magnetic-activated cell sorting (MACS) also needs large sample consumption, and specificity and affinity of the specific antibody makes it difficult to reproduce. In the field of ES cell research, single-step selective separation procedure is used for purifying undifferentiated ES cells from fibroblast feeder cells in many laboratories (Furusawa et al., 2006; Sumi et al., 2004). The method is based on the difference of adhesion affinity of the cells to a plastic dish, and it might be a less invasive method than other sorting methods using some antibodies.
Here, we obtained the fibroblast-like cells by the simple selective separation from mouse iPS cells, cultured them, and examined their MSC-like characteristics. Furthermore, we observed the differentiation properties of the cells by induction of differentiation, and demonstrated that the iPS cells can be alternative sources for cell transplantation for articular cartilage disorders.
Materials and Methods
Undifferentiated iPS culture
An iPS cell line iPS-MEF-Ng-20D-17 (Okita et al., 2007) was obtained from Riken BRC (Tsukuba, Japan). The iPS cell line was maintained on mitotically inactivated mouse embryonic fibroblast (MEF) feeder layers in Knockout-DMEM (Invitrogen Corporation, Carlsbad, CA, USA) supplemented with 20% Knockout serum replacement (Invitrogen), 2 mM L-glutamine, 0.1 mM 2-mercaptoethanol (both from Sigma-Aldrich, Inc., St. Louis, MO, USA), 0.1 mM MEM nonessential amino acid solution (Invitrogen), and supplemented with 1000 U/mL of leukemia inhibitory factor (ESGRO; Chemicon International Inc., Temecula, CA, USA). The MEF cells were prepared from C57BL/6J (SLC Co. Ltd., Shizuoka, Japan) fetuses at 13.5 days postcoitum, and used as feeder cells within three passages.
The first step of the induction of the MSC-like cells from iPS cells: Embryoid body formation and induction of EB outgrowth
In the first step of mesenchymal cell induction, we induced EB formation from iPS cells. Undifferentiated iPS cells at passage 4 were dissociated into single-cell suspension by treatment with 0.25% trypsin (Invitrogen) and 0.02% EDTA (Sigma-Aldrich) in phosphate-buffered saline (PBS) (trypsin-EDTA), resuspended in DMEM with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 0.1 mM 2-mercaptoethanol, and antibiotic–antimicotic solution (Invitrogen), and then transferred to Petri dishes. After 3 days, EBs were collected, seeded on the culture dishes at 800 to 1000 EBs/100 mm dish and cultured in DMEM supplemented with 10% FBS, 2 mM L-glutamine, and 10−6 M all-trans retinoic acid (RA, Sigma-Aldrich).
The second step of the induction of MSC-like cells from iPS cells: Collection of fibroblast-like cells from EB outgrowth
After 8 days, the outgrowths of EBs were dissociated by treatment with trypsin-EDTA. Treated cells were then filtered through 40-μm meshes (BD Biosciences Discovery Labware, Bedford, MA, USA) to remove undissociated cell aggregates. Treated cells were resuspended in 5% FBS supplemented DMEM, seeded onto the gelatin-coated dish, and cultured for 1 h. Then, the dish was washed twice to remove nonadherent cells using prewarmed medium. Adherent cells were then cultured and passaged in serum-reduced medium optimized for MSC culture, MesenPRO RS™ Medium (Invitrogen) supplemented with 2 mM L-glutamine. To promote mesenchymal cell proliferation and prevent undifferentiated cell reexpansion, we added 10 ng/mL bFGF (Upstate Biotechnology, Lake Placid, NY, USA) to the above medium.
FACS analysis for characterization of the selected cells
EB outgrowth, adherent cell fraction, and cultured adherent cells at passage 2 were used for the FACS analysis. A single-cell suspension of the cells were fixed in 2% formaldehyde in PBS for 30 min on ice, washed twice, and blocked using blocking buffer (20% FCS and 10 mM HEPES in DMEM) for 10 min on ice. Then the samples were incubated with anti-PDGFRα rabbit polyclonal antibody (diluted 1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or/and PE-conjugated anti-mouse CD105 rat monoclonal antibody (diluted 1:100; eBioscience, San Diego, CA, USA) in cold dilution buffer (PBS supplemented with 10% FCS) for 2 h on ice. The samples reacted with anti-PDGFRα antibody were washed twice and incubated with PE-conjugated anti-rabbit IgG antibody (diluted 1:500; Molecular Probes, Eugene, OR, USA) or FITC-conjugated anti-rabbit IgG antibody (diluted 1:500; Santa Cruz Biotechnology). Fluorescent intensities were observed with a FACS Caliber and the data were analyzed using Cell Quest (both from Becton Dickinson, San Jose, CA, USA). The cells reacted with PE-conjugated anti-rat IgG2a were used as an isotype control for CD105, and the cells only reacted with PE-/FITC-conjugated anti-rabbit IgG secondary antibody were used as negative control for PDGFRα.
Immunofluorescent staining of the fibroblast-like cells
The cultured adherent cells at passage 2 were fixed in Mildform 10N (Wako Pure Chemical Industries, Tokyo, Japan) at room temperature for 1 h. Fixed cells were washed with PBS and blocked by incubation in Block Ace (Dainippon Sumitomo Pharma, Osaka, Japan) for 1 h. Then the samples were washed twice and incubated with anti-PDGFRα rabbit polyclonal antibody (diluted 1:100; Santa Cruz Biotechnology) and anti-mouse CD105 rat monoclonal antibody (diluted 1:100; eBioscience) in the dilution buffer for overnight at 4°C. The samples were washed twice and incubated with Texas Red-conjugated anti-rabbit IgG antibody (diluted 1:1000; Santa Cruz Biotechnology) and FITC-conjugated anti-rat IgG antibody (diluted 1:500; Santa Cruz Biotechnology). Samples were then observed following counterstaining with DAPI diluted 1/1000 in PBS.
RNA extraction, reverse transcription, and PCR analysis
Cells were treated with TRIzol reagent (Invitrogen) and mixed thoroughly with pipetting. The Trizol-lysates were then mixed with chloroform and centrifuged at 15,000 rpm for 15 min. Following centrifugation, total RNA was obtained by isopropanol precipitation according to the manufacturer's instructions. Single-strand cDNA was prepared from total RNA using random primer under standard conditions with the High-Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). The cDNA from each sample was diluted 1:5 with RNase-free distilled water (Invitrogen) and used for RT-PCR or real-time PCR. RT-PCR amplifications were performed at 95°C for 2 min followed by 35 cycles of 94°C for 20 sec, 58°C for 20 sec, and 72°C for 20 sec using Platinum Taq PCRx DNA polymerase (Invitrogen) and appropriate primers. Amplicons were analyzed by agarose-gel electrophoresis and ethidium bromide staining. Quantitative real-time PCR with total cDNA was performed using Perfect real-time SYBR Premix Ex Taq™ II (Takara Bio, Inc., Shiga, Japan). PCR amplifications were performed with the 7700 real-time PCR System (Applied Biosystems) at 95°C for 10 sec followed by 40 cycles of 95°C for 5 sec, and 60°C for 30 sec. To quantify the relative expression of each gene, the Ct (threshold cycle) values were normalized for endogenous reference (ΔCt = Ct target – Ct β-actin) and compared with a calibrator, using the “ΔΔCt method (ΔΔCt = ΔCt sample – ΔCt calibrator)” (Dussault and Pouliot, 2006). As a calibrator, we used the average Ct value of iPS cells. Using the Ct value, relative expression was calculated (2–ΔΔCt). All experiments included negative controls consisting of no cDNA for each primer pair. All primers were designed to span exons to distinguish cDNA from genomic DNA products (Table 1).
Western blot analysis
Cells were collected by scraping, homogenized in SDS buffer (4% SDS, 125 mM Tris-glycine, 10% 2-mercaptoethanol, 2% bromophenol blue in 30% glycerol) then centrifuged at 10,000 rpm for 10 min at 4°C to remove debris. Aliquots were subjected to polyacrylamide gel electrophoresis in the presence of SDS (SDS/PAGE) followed by electrotransfer onto PVDF membrane (Hybond-P; Amersham Pharmacia Biotech, Buckinghamshire, UK). Molecular size was calibrated with Precision plus proteinTM all blue standards (Bio-Rad Laboratories, Hercules, CA, USA). The blotted membranes were blocked overnight with Block ace (Dainippon Pharmaceutical, Osaka, Japan) and treated with anti-Oct-4 rabbit polyclonal antibody (diluted 1:5000; Santa Cruz Biotechnology), anti-MMP-11 chicken polyclonal antibody (diluted 1:5000; Abcam, Cambridge, UK), anti-PDGFRα rabbit polyclonal antibody (diluted 1:5000; Santa Cruz Biotechnology), anti-Type 2 collagen mouse monoclonal antibody (diluted 1:3000; Daiichi Fine Chemical Co., Ltd., Toyama, Japan), anti-Aggrecan mouse monoclonal antibody (diluted 1:3000; Chemicon International Inc.) and anti-Actin goat polyclonal antibody (diluted 1:10,000; Santa Cruz Biotechnology) for 4 h at room temperature. Antibody incubations and washes were performed in 0.1%Tween-20 in PBS throughout. Detection was realized by enhanced chemiluminescence with an ECL plus Western blotting detection system (Amersham Pharmacia Biotech) and horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology). The lumino-labeled membranes were developed using an X-Ray Film Processor.
Osteoblast differentiation
For osteoblast induction, the cultured adherent cells at passage 4 were plated at 3 × 103/cm2 in 12-well tissue culture plate and cultured in 10% FBS-DMEM containing 100 nM dexamethasone (Sigma-Aldrich), 10 mM ascorbic acid (Sigma-Aldrich), and 10 mM 2-glycerophasphate (Sigma-Aldrich) for 10 days. Differentiated samples were tested for calcium deposition by staining with 1% alizarin red-S solution after being fixed with Mildform 10N.
Adipocyte differentiation
Adipocyte differentiation was induced by treating the cultured adherent cells at passage 4 for 10 days in DMEM/F-12 supplemented with 5% FCS and adipocyte differentiation supplement (DS Pharma Biomedical Co., Ltd., Osaka, Japan). To evaluate the adipocyte differentiation, dishes were washed twice with PBS and fixed with Mildform 10N for 1 h at room temperature. Cells were then stained for 2 h at room temperature with a filtered oil red-O solution (0.5% oil red-O in isopropyl alcohol), washed twice with distilled water, and visualized.
Chondrocyte differentiation by pellet culture
Chondrocyte differentiation property of the cultured adherent cells was examined by the pellet culture method. Pellet culture is an optimal model for the basic study of the chondrogenic differentiation capacity of the progenitors or stem cells of the chondrocyte; the products of the method can be observed histologically (Teramura et al., 2008). Cultured adherent cells (2.5 × 105) at passage 4 were centrifuged at 500 × g for 5 min and cultured as a pellet in chondrocyte differentiation medium composed of STEMPRO® Osteocyte/Chondrocyte Differentiation Basal Medium (Invitrogen) supplemented with 10% STEMPRO® chondrogenesis supplement (Invitrogen) for 3 weeks. To promote chondrogenesis, these cells were incubated under low oxygen tension (5% O2) (Domn et al., 2002; Hirao et al., 2006). The pellets were embedded in tissue-tek compound, sectioned at 6-μm thickness, and fixed for 30 min in Mildform 10N. For histological observation, fixed and washed sections were stained with safranin-O or toluidine blue. For immunofluorescent observation, the sections were blocked using Block ace for 30 min at room temperature and then incubated with anti-Type 2 collagen mouse monoclonal antibody (1:200 dilution) or anti-Aggrecan mouse monoclonal antibody (1:200 dilution) diluted in PBS contained 10% Block ace, and kept overnight at 4°C. For immunofluorescence microscopy, the samples were incubated for 1 h at room temperature with FITC-conjugated anti-mouse IgG antibody (diluted 1:1000; Santa Cruz Biotechnology). These samples were counterstained with DAPI before observation.
Evaluation of the chondrocyte differentiation properties of the induced cells
To analyze chondrocyte-specific gene expression or protein expression, we used micromass culture with 96-well culture plate (Penick et al., 2005). Micromass culture is an assay system that mimics the first step of mesenchymal condensation in normal cartilage development.
To induce chondrocyte by the micromass culture method, the cultured adherent cells at passage 4 were seeded at high density of 2.5 × 105 cells per well in a 96-well plate precoated with 0.1% gelatin. After incubation for 24 h, 200 μL of the chondrocytes differentiation medium was carefully added to each well and cultured for 14 days in a humidified atmosphere of 5% O2, and 5% CO2 at 37°C. Medium change was performed every alternate day.
For a control experiment, unselected EB outgrowth cultured for 8 days in DMEM supplemented with 10% FBS, 2 mM L-glutamine, and 10−6 M RA and mouse chondrocyte progenitor cell-line ATDC5 (Riken BRC) at passage 4 were induced to differentiate into chondrocyte at the same conditions as above.
Statistical analysis of the data
A significant difference was detected by the Tukey-Kramer HSD test or Student's t-test. A p-value of less than 0.05 was considered significant.
Results
Generation of fibroblast-like mesenchymal cells from iPS cells
To obtain the cells possessing the properties of mesenchymal progenitors, we first produced mesenchymal lineage-committed cells via EB formation and derivation of outgrowth of the EBs (Fig. 1A). Furthermore, we used RA as an inducer of the differentiation because it was reported that the MSCs could be induced from mouse ES cells in RA-added condition (Takashima et al., 2007). To examine the appropriate concentration of RA for the differentiation of iPS cells, we cultured the EBs in various RA concentrations for 1 week. When the EBs were cultured in low RA concentration (10−8–10−7M) conditions, reexpansion of undifferentiated cells were observed in some cases, and pluripotent state-specific gene expressions were observed within the next series of differentiation (data not shown). In the presence of 10−6 M RA, reexpansion of undifferentiated cells was prevented, whereas the expression of an important mesenchymal progenitor cell marker PDGFR α (Betsholtz, 2004; Takashima et al., 2007) was maintained (Fig. 1B).
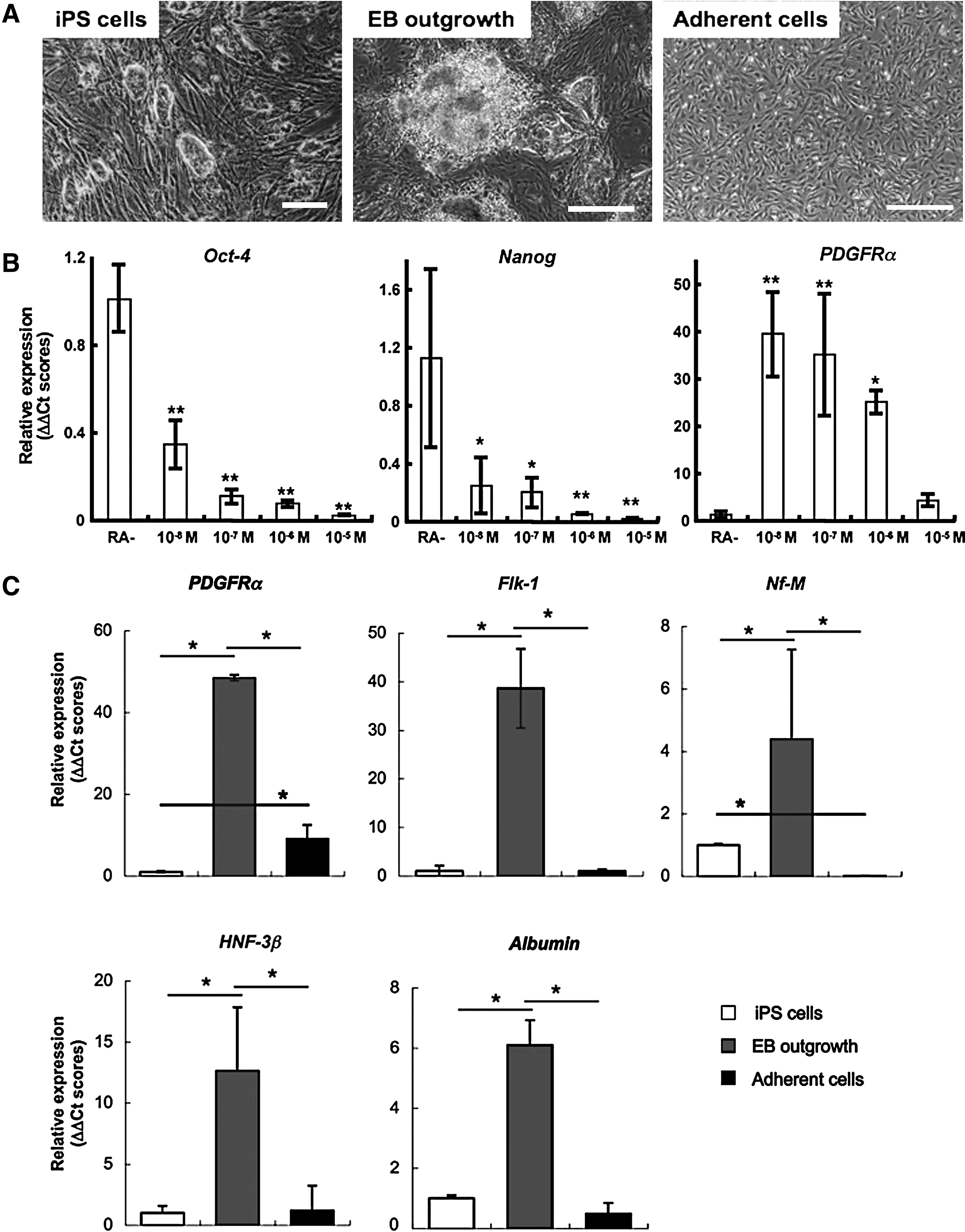
Derivation of mesenchymal lineage-committed cells by EB formation. (
When performing induction of the outgrowth from EBs in the above condition, elevation of multiple differentiation marker gene expressions including PDGFRα was observed (Fig. 1C). Immunofluorescent observation elucidated that the PDGFRα-expressing putative mesenchymal cells showed fibroblast-like morphology, and these cells were mainly in the outer area of the EB outgrowth (Fig. 2A).
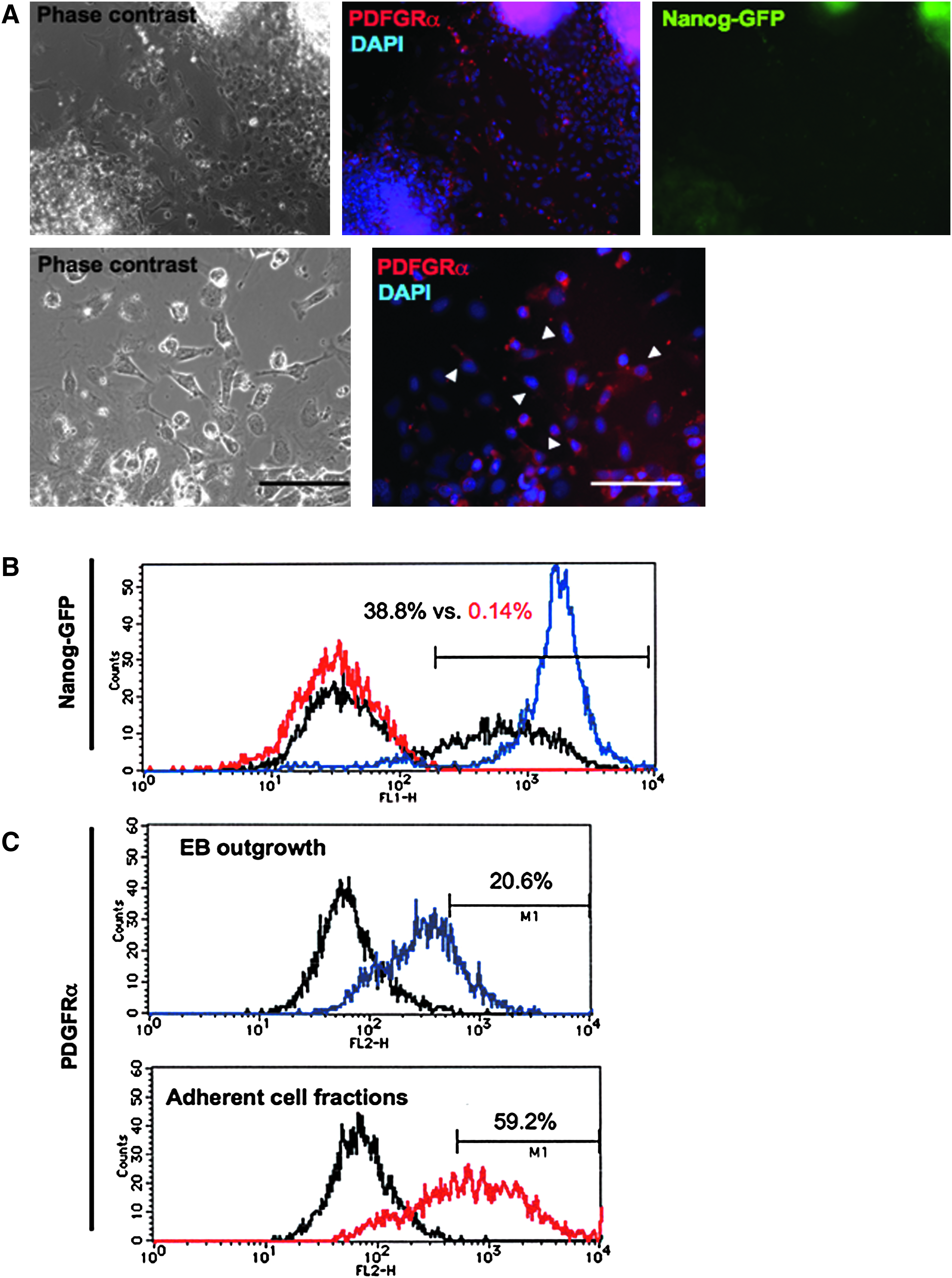
Immunofluorescent observation of the mesenchymal lineage-committed cells. (
In the next experiment, we select the fibroblast-like cells using different adherence properties of the cells from other types of cells such as neural cells, epithelial cells, or undifferentiated cells. After the selection procedure, that is, enzymatic digestion, short-time culture, and removing unadherent cells, approximately 10% of total cells were collected as an adherent cell fraction, and only fibroblast-like cells could expand in the following culture. The fibroblast-like cells showed approximately 10-fold higher expression of PDGFRα than undifferentiated iPS cells, although other lineage markers such as Flk-1 (epithelial cell), Nf-M (neural cell), Hnf-3β (endoderm), and Albumin (hepatocyte) did not increase (Fig. 1C).
Then we evaluated the purity of the collected cells by FACS analysis for GFP expression and lineage specific markers. The iPS cells used in the present study contain the GFP reporter gene that is controlled by the Nanog promoter (Nanog–GFP; Okita et al., 2007), so the cells at the undifferentiated state express GFP fluorescence. In the culture of EBs and the outgrowth, GFP-expressing undifferentiated cells remained in the center of the EBs (Fig. 2A). FACS analysis showed approximately 40% of total cells representing GFP expression. The rate of the GFP-expressing cells dramatically decreased to about 0.1% in the reexpanded fibroblast-like cell culture at 48 h after the separation (Fig. 2B). On the other hand, rate of the PDGFRα-expressing cells increased about threefold higher than before the selection (Fig. 2C). By the following culture in the medium optimized for MSC culture, rate of the PDGFRα-expressing cells further increased, over 80% of total cells expressed PDGFRα and approximately 60% of total cells expressed an MSC marker CD105 (Fig. 3A).
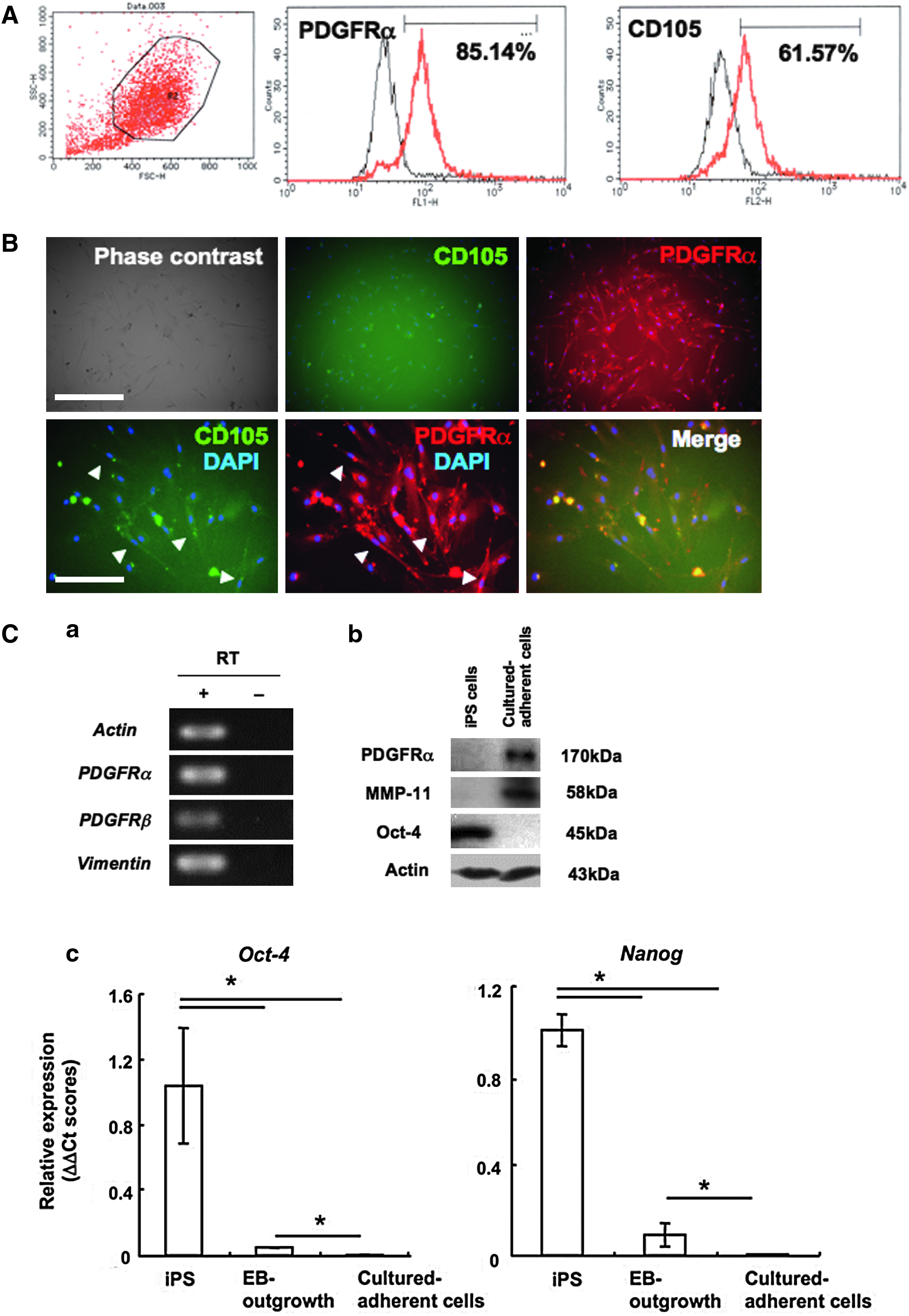
Detailed analysis of the cultured adherent cells to examined their MSC-like properties. (
Detailed characterization of the fibroblast-like cultured adherent cells
To characterize the fibroblast-like cells, we performed immunofluorescent staining, real-time PCR and Western blot analysis for mesenchymal lineage markers or MSC-specific markers. Immunofluorescent staining revealed that almost all cells expressed PDGFRα, and mainly thin spindle-shaped cells coexpressed CD105 (Fig. 3B). RT-PCR analysis showed that the cells expressed other mesenchymal cell markers PDGFRβ and vimentin, and MSC markers CD29, CD34, CD106, and Bmi-1 (Figs. 3C-a and 4B). PDGFRα expression and fibroblast cell marker MMP-11 expression were determined by Western blot analysis (Fig. 3C-b). On the other hand, expressions of pluripotent cell markers Oct-4 and Nanog were dramatically reduced (Fig. 3C-c). In the present study, bFGF supplementation significantly improved proliferation of the fibroblast-like cells. These cells could be maintained and passaged six times in the condition (Fig. 4A). The property to respond to bFGF supplementation is also evidence that the cells induced in the present protocols possessed mesenchymal cell character (Schmidt et al., 2006). However, the proliferation property was limited, and expansion of the cells over seven time passages was difficult. Expressions of stem/progenitor markers CD34 and Flk-1 gradually decreased by passages (Fig. 4B and Supplementary Fig. 1).

Self-renewal properties of the cultured adherent cells. (
In the next series of the study for characterization of the cells, we observed differentiation properties of the fibroblast-like cells. When the cells were transferred to osteogenic condition, a high rate of osteoblast-committed cells were observed. Calcium deposition was determined by alizarin red staining (Fig. 5). Moreover, when the cells were cultured in the adipogenic condition, some part of cells differentiated to adipocytes. Lipid droplet accumulation was examined by microscopy after staining with oil red-O (Fig. 5). Then we cultured the fibroblast-like cells as a pellet and evaluated their chondrogenic properties. After 3 weeks of culture, the pellet composed of chondrocytes surrounded by proteoglycan-rich extracellular matrix was obtained. The cell density was higher in the peripheral part than in the central, and intense safranin-O staining was present mainly in the peripheral regions. These areas were stained by toluidine blue to red-purple (metachromatic staining). Immunofluorescent staining with specific antibodies to Aggrecan and Type 2 collagen more clearly demonstrated that chondrocytes with abundant cartilage-specific matrix existed in the pellets (Fig. 5).
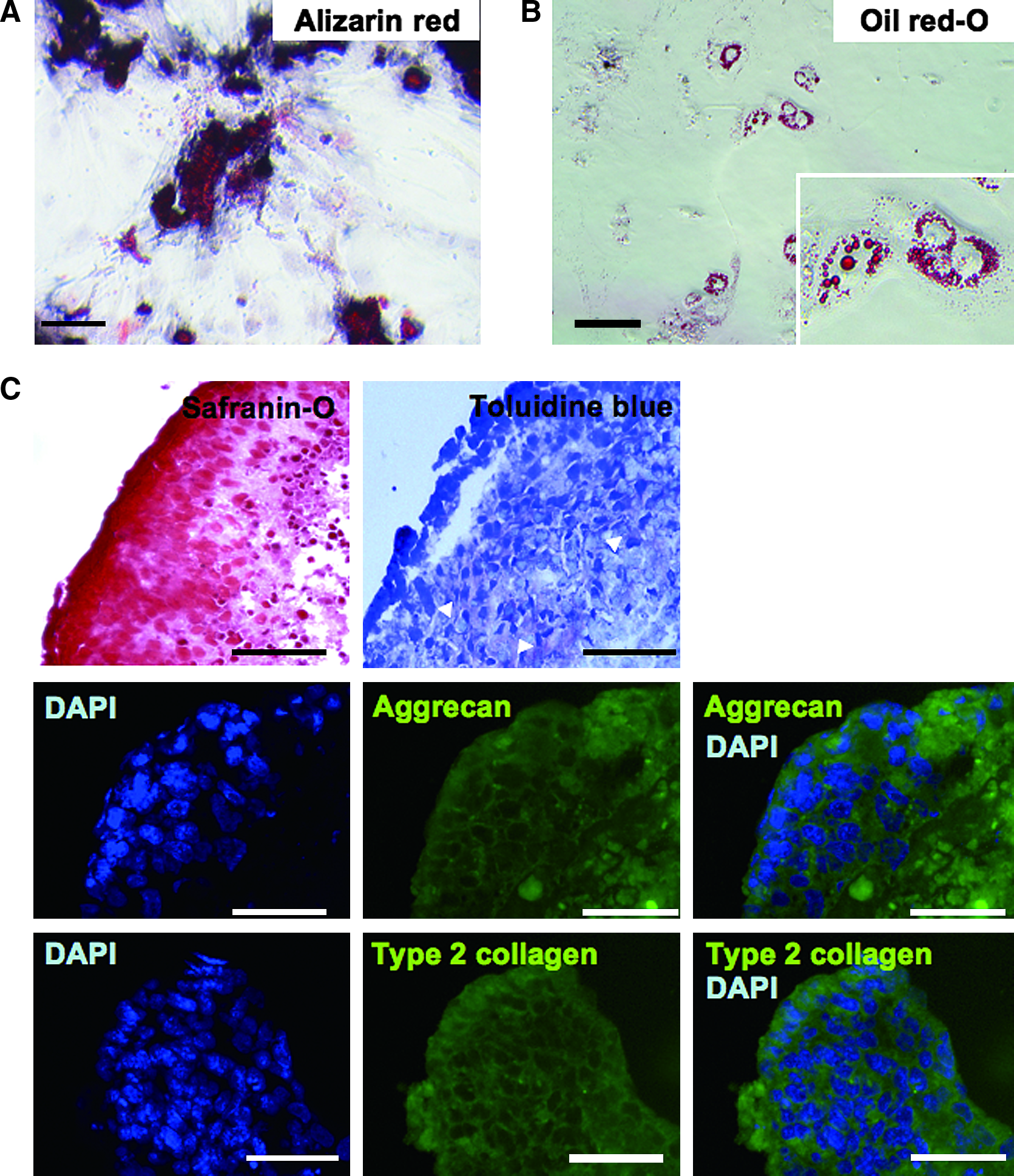
Differentiation assays for the cultured adherent cells. The cultured adherent cells maintained multiple differentiation properties to the osteoblast, adipocyte, and chondrocyte. (
From these characteristics, we concluded that the fibroblast-like cells obtained by the present protocol possessed important and specific properties as MSCs, and then we defined the cells as iPS–MSCs.
Evaluation of iPS–MSC-derived chondrocyte
To quantify and evaluate the characteristics of the iPS–MSC-derived chondrocyte, we performed induction of differentiation by micromass culture. After 14 days, the differentiated cells showed chondrocyte-specific gene expression properties. Sox-9, a master transcription factor regulates the gene expression of chondrocyte matrix proteins, and the Col2A1, a chondrocyte-specific extra cellar matrix, significantly increased after differentiation. By differentiation induction, Sox-9 expression was up to threefold from iPS–MSC and approximately an 80-fold increase was observed when compared with that of undifferentiated iPS cells. Col2A1 expression also increased after differentiation induction. The expression quantity was enhanced about 20-fold higher in chondro-differentiated iPS–MSCs from the preinduced state (Fig. 6A). Chondrocyte-specific matrix expressions were also determined by Western blot. The clear expression of Type 2 collagen and Aggrecan were detected in chondro-differentiated MSC-like cells and ATDC5. On the other hand, the expression of pluripotent marker Oct-4 was not detected in iPS–MSC and iPS–MSC chondrocyte; it was only observed in undifferentiated iPS cells and the EBs (Fig. 6B). These results clearly demonstrated that the method that the selective expansion of progenitors before inducing the final product was effective in avoiding the risk of contamination of the undifferentiated iPS cells, at least in the case of chondrocyte differentiation.
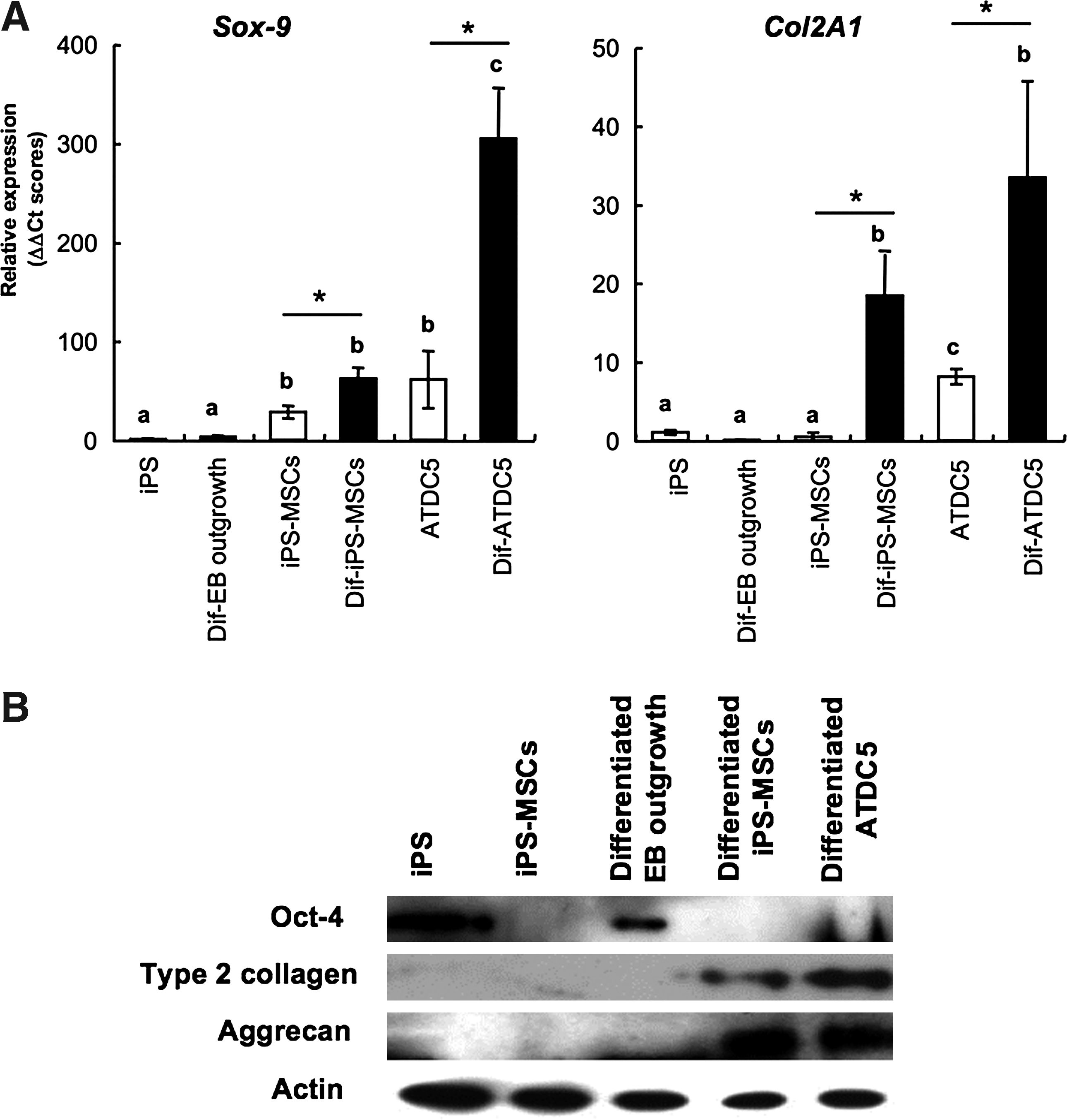
Chondrocyte-specific gene expressions after induction of differentiation by the micromass culture. Each gene expression was determined by real-time PCR (
Discussion
One of the major problems in the clinical application of the pluripotent cells such as ES cells or iPS cells is that the final product for transplantation often will be a heterogeneous population with contamination of undifferentiated cells. In the present study, we suggested the simple separation method to obtaining the cells showing MSC-like characteristics from mouse iPS cells.
By selective separation of adherent cells, PDGFRα-expressing candidate cells were effectively collected, and Nanog–GFP-positive undifferentiated cells dramatically reduced. The culture of the adherent cells in the optimized medium for MSCs might also function as further selection of the iPS-derived MSC-like cells. The FGFs such as bFGF (FGF-2) and FGF-4 are the major stimulus activating Erk and important mitogen for MSCs (Schmidt et al., 2006). Moreover, bFGF also decrease the risk of contamination of undifferentiated pluripotent cells because the Erk elevation by FGF-addition inhibit mouse ES cell pluripotency (Kunath et al., 2007). Addition of the bFGF significantly promoted the expansion property of the iPS-derived MSCs when compared with the control condition without bFGF. However, the cells lost the proliferation properties around six passages, and expressions of stem/progenitor cell markers CD34 and Flk-1 declined.
We think the decline of the self-renewal property of the iPS–MSC was probably due to the following two reasons. First, the genetic background of the iPS–MSCs may strongly affect the capacity. It was reported that the characteristics and culture conditions of mouse bone marrow-derived MSCs were considerably different between mouse strains. Furthermore, Peister et al. (2004) reported that it was difficult to expand the MSCs derived from GFPtg strains. The iPS cells used in the present study contained some transgenes, including GFP, and it might be one of the major reasons for low proliferation property of the cells. The second point is the differentiation condition optimized for iPS cells. Recently, Takashima et al. (2007) reported that MSC-like cells possessing longer expansion capacity could be induced from mouse ES cells. They used 10−7 M RA as an inducer for differentiation. When we used RA at 10−7 M for differentiation induction of iPS cells, undifferentiated cells remained in some cases. Morizane et al. (2009) also observed difficulty of differentiation in iPS cells. They suggested that transfected exogenous factors strongly suppress the differentiation, and result in cells remaining undifferentiated cells. In the present study, we determined that undifferentiated cells could be eliminated by increasing RA concentration to 10−6 M. However, high concentration RA leads to negative side effects. It was reported that 1 μM (10−6 M) RA strongly hampers the growth of bone marrow-derived MSCs in vitro (Oliva et al., 2003).
To improve the self-renewal capacity of the iPS–MSCs, further optimization of the medium for iPS–MSC expansion and the condition of differentiation induction, are necessary as well as a detailed study for the mechanisms of limiting the self-renewal of iPS–MSCs by conducting comparative studies with iPS cells generated by the introduction of a reduced number or absence of the transgenes for reprogramming factors (Kim et al., 2009; Zhou et al., 2009).
When the iPS–MSCs were used for the induction differentiation, the levels of chondrogenesis related genes Sox-9 and Col2A1 significantly increased. In contrast, these gene expressions hardly increased when EB outgrowth was used for chondro-differentiation in the control study. Tissue-specific progenitor cell microenvironments are required for chondrogenesis and cartilage formation in the developing limb. These environments may include specific cell–cell interactions, cell–matrix interactions, and appropriate cytokine signals such as bFGF, TGF-β, BMP-2, 4, and PDGF. These cytokine signals possess positive effects in chondrocyte differentiation; chondrocyte maturation, and cartilage-specific–extracellar matrix production was promoted when they act in an appropriate combination. However, it was also reported that addition of inappropriate timing or combination of the cytokines might result in inhibition of chondrocyte differentiation or decreasing matrix production (Kramer et al., 2000; Nakayama et al., 2003; Ogawa et al., 2003). EB outgrowth contained various kinds of cells, and these cells were attached to each other. Furthermore, it was possible that some growth factors were expressed around the progenitors of chondrocyte. These situations might result in the lower efficiency of chondrocyte differentiation from EB outgrowth. In contrast, purified iPS–MSCs might not be affected from these negative factors and could differentiate to chondrocyte successfully.
Interestingly, the MSC-like cells expressed Sox-9 before induction of chondrocyte differentiation. It was reported that chondrocyte-committed cells could be observed by only transferring the ES-derived EBs to adherent condition (Hegert et al., 2002; Kramer et al., 2005; Hargus et al., 2008). Also in the present study, the chondrocyte-committed cells might be included in the adherent fibroblast-like cell fractions. Increase of Sox-9 expression by chondro-differentiaion was significantly smaller than that of ATDC5, presumably because of the difference in the developmental stages of both cells. ATDC5 is a mouse chondroprogenitor cell line, and it easily transmits to mature chondrocyte by differentiation induction. On the other hand, MSC is a stem cell of mesenchymal lineages including osteocyte, myocyte, adipocyte, and chondrocyte. It was supposed that the MSCs are in less mature state than ATDC5, so that the increase of Sox-9 expression was lower than ATDC5.
To promote chondrocyte formation from stem cells and to elevate gene expressions of cartilage specific matrix, we cultured the MSC-like cells in hypoxic condition. The positive effect of the hypoxia in chondrocyte differentiation relates to the regulation of hypoxia-inducible factor family (HIFs) (Koay and Athanasiou 2008). HIFs are stabilized under hypoxic condition, and function as transcription factors of various genes. In chondrocytes, it was reported that one of the HIF family members, HIF-2α, induced Sox-9 expression (Lafont et al., 2007). In addition to the promotion effect of chondrocyte differentiation, it was reported that HIF-1α, which is another member of HIF, inhibits mouse ES cell self-renewal via repression of LIF receptor (Jeong et al., 2007). Furthermore, Powers et al. (2008) described that hypoxic conditions of 0 to 5% O2 progressively decreased the expressions of the pluripotent related genes Oct-4, Nanog, and Sox-2 when mouse ESCs were transferred into differentiation condition. Also in the present study, we confirmed same tendency, that is, Nanog and Sox-2 or a tumor formation related gene E-ras was down-regulated when undifferentiated iPS cells were cultured in hypoxic condition (a part of the data is shown in Supplementary Fig. 2B). In human pluripotent cells, hypoxic condition of 5% O2 promotes self-renewal and enhances pluripotent state-related gene expressions but severe hypoxic conditions such as 1% O2 inhibit survival of the undifferentiated cells (Prasad et al., 2009). On the contrary, it was reported that the severely hypoxic condition is favorable for chondrocyte to present their characteristics (Lafont et al., 2007), therefore culturing the iPS-derived MSC-like cells in hypoxic condition at the final step of differentiation might be effective in reducing the risk of contamination by undifferentiated cells and might be a useful technique in providing the transplantable chondrocyte.
Here, we first demonstrated that the cells possessing MSC-like characteristics were obtained from mouse iPS cells. Although further improvements of differentiation conditions or detailed functional study such as transplantation to animal models are essential in developing a stronger protocol for preparing higher quality cells, our approach provides important evidence to show the possibilities of iPS cells to become an alternative source for regenerative medicine in articular disorders.
Footnotes
Acknowledgments
This study was supported by grants from the Nakatomi Health Science Foundation and the Grant-in-Aid for Young Scientists (startup, grant number 20890263) from the JSPS. We gratefully acknowledge Ms. Naomi Backes Kamimura, Department of Biology-Oriented Science and Technology, Kinki University, for English editing.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.