
Abstract
Abstract
It has been demonstrated that several types of somatic stem cells have the remarkable capacity to differentiate into other types of tissues. However, the promise of keratinocyte stem cells seems slim for generating nonepidermal tissues. Using our recently developed acclimatization induction strategy, we demonstrate the multipotency of adult human undifferentiated keratinocytes (UKs). The UKs were isolated from the basal layer of adult human foreskin and cultured in Epilife medium, which allows for the growth of only keratin-positive keratinocytes, promotes high proliferation of UKs, and prevents their differentiation. Induction of the UKs by either serum or lineage-committed medium only produce differentiated epidermal cells. Hence, serum or lineage-committed medium was added to Epilife to acclimate UKs to differentiate to other cell types. Unexpectedly, serum acclimatization can induce UKs to produce a large number of smooth muscle cells and fewer of adipocytes and neurocytes within 3 weeks. In contrast, except for the terminally differentiated epidermal cells, committed acclimatization can induce UKs to differentiate exclusively into the adipocytic, myogenic, or neurogenic lineages. These data indicate that human UKs represent a novel multipotent adult stem cell, and suggest that they may provide an accessible, therapeutically promising cell source for regenerative medicine.
Introduction
The most obvious therapeutic use of such multipotent adult human precursors is for cell transplantation and replacement. In such case, the ideal human precursor cell population would be one that could be derived in an autologous fashion from small amounts of accessible human tissue. However, the difficulty in reliably generating large numbers of stem cells from adult humans hinders clinical translation of these therapeutically promising cells. Skin is the largest organ of the human body and the epidermis, of which is a highly accessible tissue source. An emerging view of epidermal stem cells is the existence of at least three distinct niches for skin stem cells: the follicle bulge, the base of the sebaceous glands (SG), and the basal layer of the epidermis (Blanpain and Fuchs, 2009; Fuchs, 2007; Fuchs and Horsley, 2008). Among these, the basal layer cells of epidermis are abundant and enriched with keratinocyte stem cells. These undifferentiated keratinocytes (UKs) in mammalian skin are readily accessible and easy to isolate and expand in vitro (Li et al., 1998; Lichti et al., 2008; Papini et al., 2003). A previous study showed that the epidermal stem cells isolated from the mouse basal-layer epidermis could differentiate into multiple cell lineages during development when they were injected into blastocysts, suggesting that UKs could be a potential stem cell source for regenerative medicine (Liang and Bickenbach, 2002). However, whether resident progenitor cells in the basal layer of epidermis can be induced to differentiate into other lineages remains unknown (Fuchs, 2007). Here we describe our use of an acclimatized induction approach to investigate the plasticity of adult human UKs.
Materials and Methods
Cell culture
Pieces of human foreskin of 15–22 cm2 deriving from voluntary circumcisions of adult aged 18 to 49 years of age were washed with cool phosphate-buffered saline (PBS) (Gibco, Grand Island, NY, USA). The method of keratinocyte isolation from the epidermal basal layer was mainly as described previously (Kim et al., 2004; Li et al., 1998; Papini et al., 2003), with modification. Briefly, an epidermal sheet was obtained from each sample, and isolated epidermal cells were seeded at approximately 2 × 106 cells per well in a six-well culture plate coated with collagen IV (Sigma-Aldrich, St. Louis, MO, USA). After a 1-h incubation at 37°C, unattached cells were gently removed by aspiration, and attached keratinocytes were then maintained in serum-free and low-calcium EpiLife medium supplemented with HKGS (Cascade Biologics, Portland, OR, USA). The growth medium was completely changed every 2 to 3 days. Typically, cells at 80–90% confluence were passaged. The skin fragments were obtained after the patients were informed about the purpose for which the skin was collected, agreed, and signed an informed consent form. This study was approved by the Ethics Committee of Chinese PLA General Hospital.
The ventricular tissue of a beating heart from a neonate Sprague-Dawley rat (1 day old) was sliced with scissors into a small piece of 0.5 to 1 mm3. Myocardial cells isolated with use of trypsin and collagen II digestion were cultured in Dulbecco modified Eagle medium (DMEM) (Gibco) containing 10% fetal bovine serum (FBS) (Gibco) in an incubator with 5% CO2 at 37°C. The medium was refreshed every 3 days.
Differentiation induction
UKs at 50–90% confluence and at passages 1 to 5 were mainly used for noncommitted serum induction, lineage-committed adipogenic, and myogenic differentiation induction in six-well plates. DMEM-F12 (Gibco) containing 10, 5, or 2% FBS was used for serum induction differentiation. IMDM (Gibco) containing 10% FBS and isobutyl-methylxanthine (0.5 mM), dexamethasone (1 μM), insulin (0.5 mM), and indomethacin (0.2 mM) were used for adipogenic differentiation (Pittenger et al., 1999). The supernatant of cultured mouse myocardial cells and retinoic acid (RA, 0.5 μM) was used for myogenic differentiation of UKs.
UKs at 80–95% confluence were used for neurogenic differentiation. Cells were first stimulated by adding 0.5 mM β-mercaptoethanol (ME), 45 ng/mL basic fibroblast growth factor (bFGF) (Invitrogen, Carlsbad, CA), 20 ng/mL epidermal growth factor (EGF; Invitrogen), 0.5 mM RA, 0.5 mM valproic acid (VPA), 2000 U/mL leukemia inhibitory factor (LIF) (Millipore, Billerica, MA, USA), 100 mM L-glutamine, 30 mM glucose, 5 μg/mL insulin, 1 mM sodium pyruvate, and 2% DMEM-F12 (containing 2% FBS) into EpiLife medium for 36 h. Cells were then cultured for 2 days in medium with 20% DMEM-F12 (containing 2% FBS) and 80% EpiLife containing 20 ng/mL bFGF, 20 ng/mL EGF, 0.5 μM RA, 1 × B27 (Invitrogen), and 100 μg/mL butylated hydroxyanisole (BHA). Finally, the amount of DMEM-F12 (containing 2% FBS) was increased to 100% at a 20% increase every 2 days. All chemicals were purchased from Sigma Aldrich unless otherwise indicated.
Flow cytometry analysis
A total of 5 × 105 freshly isolated or cultured keratinocytes (3–5 day) were suspended in 100 μL cool PBS and incubated with 10 μL fluorescein isothiocyanate (FITC)-labeled CD90, CD24, β1 integrin, CD34, CD133 monoclonal antibodies, or FITC-mouse IgG1a isotype antibody (all from BD Biosciences, San Diego, CA, USA) on ice for 30 min, then were washed three times with PBS and resuspended in 500–800 μL PBS. Hoechst 33528 (Sigma-Aldrich) was included at 1 μg/mL in the final wash to detect dead cells. For the immunophenotype characterization, a total of more than 10,000 events were collected and were analyzed by FACSCalibur (BD Biosciences).
Reverse-transcription-polymerase chain reaction (PCR)
Total RNA was extracted by use of TRIzol reagent (Invitrogen), and cDNA was generated with 1 to 2 μg total RNA and Superscript II RNase H− reverse transcriptase (Invitrogen) as described by the manufacturer. Oligonucleotide primers were PPARγ2 sense, 5′-ATGACAGCGACTTGGCAATA-3′ and antisense, 5′-GCAACTGGAAGAAGGGAAAT-3′ (341 bp); LPL sense, 5′-TCGTTCTCAGATGCCCTACAA-3′ and antisense, 5′-TTCCTCCACTTCATTCTTCAC-3′ (496 bp); C/EBPα sense, 5′-GAACAGCAACGAGTACCGGGTA-3′ and antisense, 5′-CCATGGCCTTGACCAAGGAG-3′ (224 bp); MyoD sense, 5′-CCAGCGGTTGCCCAAGGTG-3′ and antisense, 5′-GACCCCACCCAGTCACCGGA-3′ (483 bp); Myf5 sense, 5′-ACGACCAACCCCAACCAGAGG-3′ and antisense, 5′-TAGTGGACCGGATCACCTCCTCA-3′ (321 bp); Sm22α sense, 5′-GGTGAAGGTGCCCGAGAACCCA-3′ and antisense, 5′-ATCTGCCGAGGTCCGTAGC-3′ (369 bp); nestin sense, 5′-TTGGAACAGAGGTTGGAGGGC-3′ and antisense, 5′-TCTCAAGGGTAGCAGGCAAGG-3′ (373 bp); β-actin sense, 5′-AAAGACCTGTACGCCAACAC-3′ and antisense, 5′-GTCATACTCCTGCTTGCTGAT-3′ (219 bp).
Immunocytochemistry and quantification
Cells on chamber slides were washed three times with PBS and fixed with 4% paraformaldehyde for 15–20 min. Cells were permeabilized with 0.5% Triton X-100 for 20 min, then blocked for 1 h at room temperature with 1% bovine serum albumin and 6% normal goat serum in PBS. Primary antibodies were then added in PBS containing 3% normal goat serum and left overnight at 4°C. Primary antibody was removed, cells were washed three times for 5 min with PBS, and the appropriate secondary antibody conjugated to Cy3 or FITC was added in PBS containing 3% normal goat serum for 1 h at room temperature. Nuclei of the cells were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich). Immunofluorescence analysis was performed using a Nikon fluorescence microscope equipped with the appropriate filters for three-color imaging of cells with a CDD camera. The primary antibodies used were antinestin monoclonal (1:200), anti-β3-tubulin monoclonal (1:200), anti-GFAP polyclonal (1:200, DAKO, Copenhagen, Denmark), anti-SMA monoclonal (1:400), anti-CK14 polyclonal (1:400), anti-CK5 polyclonal (1:400), anti-β1 integrin polyclonal (1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA), antiinvolucrin polyclonal (1:350, Santa Cruz Biotechnology), anti-c-Kit monoclonal (1:300, Santa Cruz Biotechnology), and antifibronectin polyclonal (1:200). Secondary antibodies were Cy3-conjugated goat antimouse (1:400–600), Cy3-conjugated goat antirabbit (1:400–600), FITC-conjugated goat antimouse (1:400–600), and FITC-conjugated goat antirabbit (1:400–600). Antibodies were purchased from Sigma-Aldrich unless otherwise indicated.
For quantification of the percentage of cells producing a given marker protein, at least three fields were photographed in each experiment, and the number of positive cells relative to the total number of DAPI-labeled nuclei was determined. In a typical experiment, each field contained 100 to 500 cells, and a total of 500 to 1500 cells were counted per marker.
Karyotyping analysis
Standard R-banding chromosome analysis was performed in the Cytogenetics Lab of Hematology Department in Chinese PLA General Hospital. Briefly, keratinocytes were cultured as described above until near confluency, the colcemid (Gibco) was added to the culture with a final concentration of 0.1 μg/mL and incubated for another 4 h. Then the cells were harvested, treated with hypotonic solution (0.0075M KCl) at 37°C for 45 min, and fixed by fresh Carnoy's solution (3: 1 = methanol:acetic acid). R-banding was performed by staining the slide in 10% GIMSA solution for 10 min. A total of 20 metaphases were analyzed for each sample under Nikon E800 microscope.
Results
UKs can be routinely isolated and expanded from adult human foreskins
Adult human foreskin tissue was used in our attempts to isolate and characterize human UKs. Foreskin samples from circumcisions of adult ranging from 18 to 49 years of age were used for this study, with over 100 samples in all being analyzed. These samples were indistinguishable in terms of UKs isolation and properties.
To isolate UKs, epidermis and dermis were dissociated from foreskin samples of about 15–22 cm2, and isolated epidermal cells (9 × 106 to 1.2 × 107 for each sample) were seeded at approximately 2 × 106 cells per well in a six-well culture plate coated with collagen IV. It has been reported that the cells adhering to collagen IV within 1 h are enriched with UKs from the basal layer (Dong et al., 2007; Kim et al., 2004). So we removed the unattached cells after 1 h and cultured the attached keratinocytes in EpiLife medium, which allows more than 95% cell confluence being reached after 3–5-day culture. CD90, CD24, and β1 integrin are well-documented markers of UKs (Janes et al., 2002; Nakamura et al., 2006; Piwko-Czuchra et al., 2009; Redondo et al., 1998). By comparison with those in freshly isolated cells, the proportion of cells expressing CD90, CD24, and β1 integrin in 3–5-day cultured keratinocytes were obviously increased (Fig. 1A). Consistent with the previous reports (Aasen et al., 2008; Zhang et al., 2009), these results demonstrated that Epilife medium allows for the growth of only keratin-positive keratinocytes, promotes high proliferation of UKs, and prevents their differentiation. Unlike UKs from junior foreskin samples reported in earlier study (Aasen et al., 2008), adult UKs exhibited slightly low expression profiles for CD90 (21.7 ± 3.8%), CD24 (83.4 ± 6.2%) and β1 integrin (83.3 ± 12.1%). These differences may suggest differential UK proportion existing in junior and adult samples. Certainly, we did not exclude that these differences were caused by different research systems. In addition, we measured the expression of CD34 and CD133 in 3–5-day cultured cells by flow cytometry. The CD34 antibody has been shown to delineate cells in the outer root sheath of the human hair follicle but not epidermal basal layer cells (Poblet et al., 1994). CD133 labels epithelial stem cells in the prostate (Richardson et al., 2002) but only one isoform AC133-2 has been shown to be expressed in cultured keratinocytes (Yu et al., 2002). Consistent with most of these previous reports, we did not detect CD34 and CD133 expression in cultured keratinocytes isolated from foreskin epidermis (Fig. 1A).

Culture and identification of adult human UKs isolated from the basal layer of adult human foreskin epidermis. (
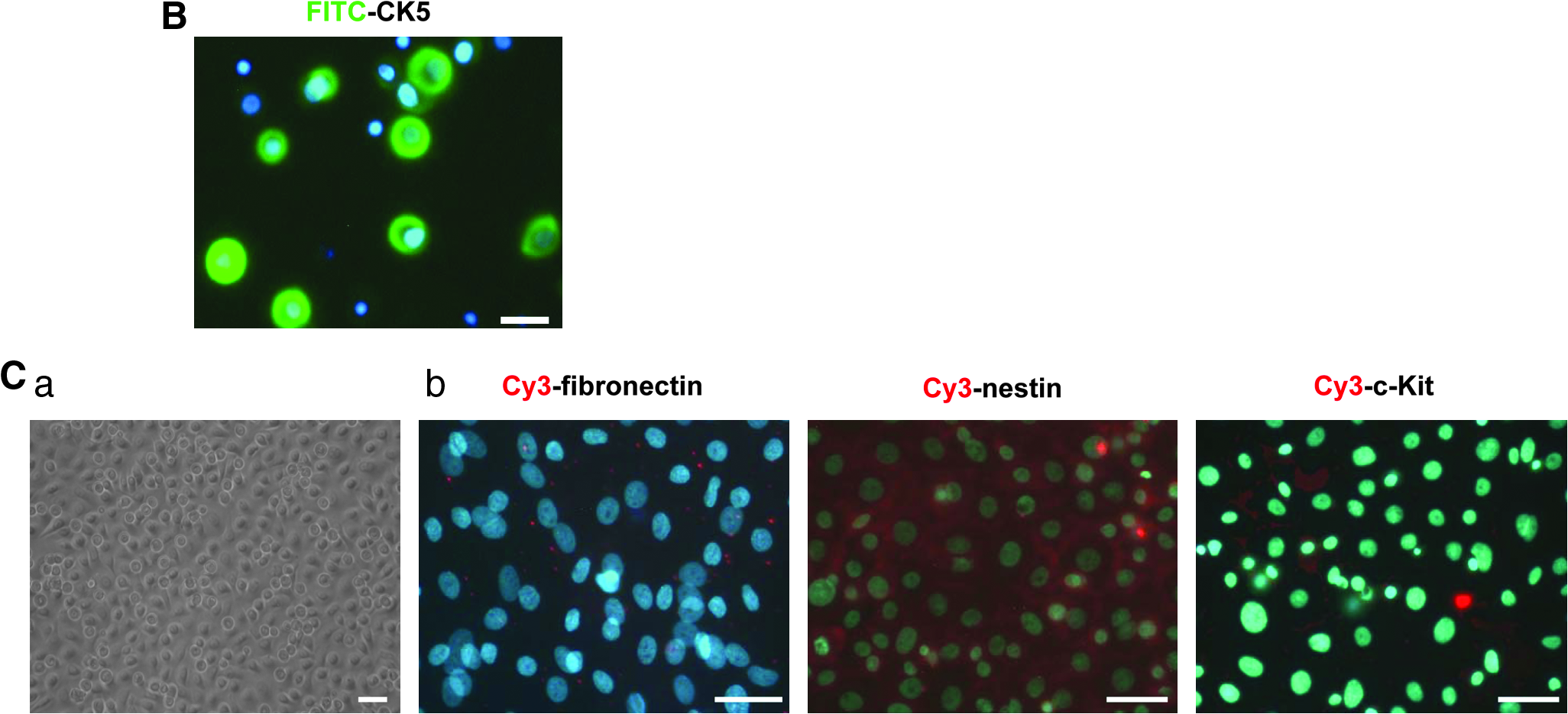
The basal to suprabasal (or spinous) transition is a key first step in the program of terminal differentiation of UKs. As cells enter the spinous layer, they switch off the expression of genes encoding cytokeratin 5 (KRT5; also known as CK5) and CK14 (Blanpain and Fuchs, 2009). By immunofluorescence analysis, we observed that more than 50% isolated keratinocytes (within passage 1) were positive for both CK5 and CK14 expression in this culture system (Figs. 1B and 6A).
During the culture period of UKs, no subpopulations of melanoblasts, dermal fibroblasts, or dermis-derived stem cells could be discerned morphologically by microscopic observation. To further exclude the contamination of these cells, we performed immunofluorescence analysis using anti-c-kit, antifibronectin, and antinestin antibodies, which are well-documented molecular markers of melanoblasts, dermal fibroblasts and dermis-derived stem cells, respectively. As shown in Figure 1C, the c-kit-, fibronectin-, or nestin-positive cells were undetectable in this culture system.
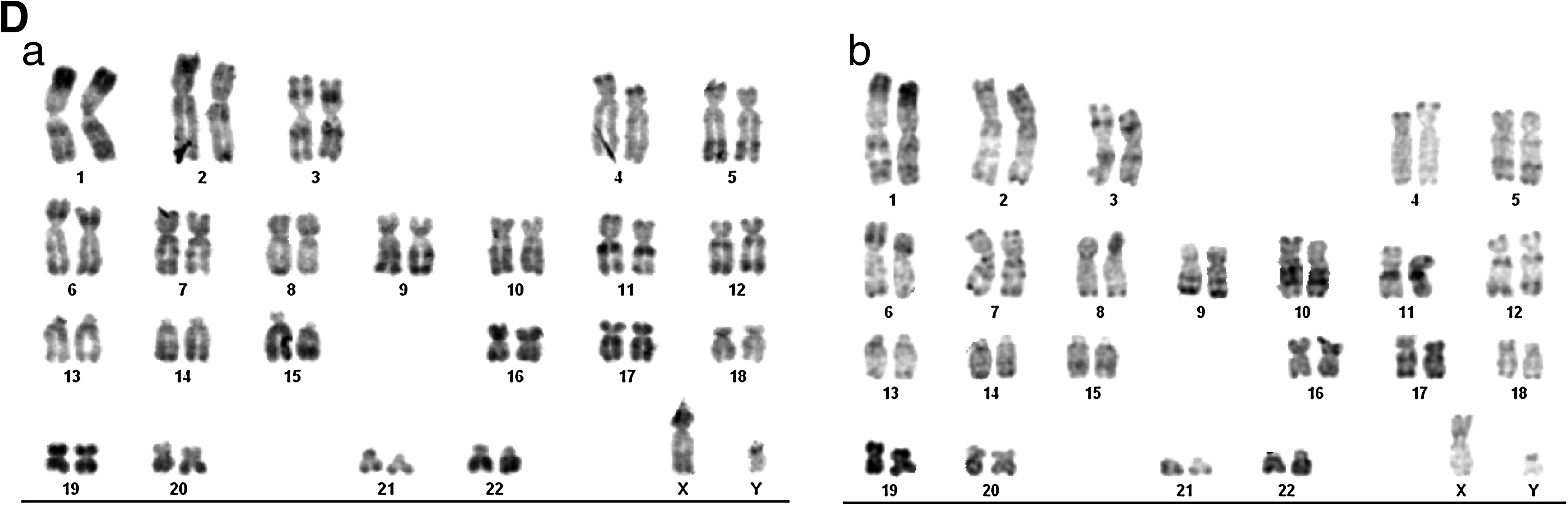
Finally, we performed karyotyping analysis in cultured keratinocytes. As shown in Figure 1D, R-banding of keratinocytes from one sample demonstrated that the chromosomal karyotypes were normal in both early-passaged cells (passage 1) and late-passaged cells (passage 6).
UKs generates mesodermal progeny and nestin-positive cells by serum acclimatization induction
Certain adult stem cells, such as those derived from the dermis of mammalian skin, manifest their multipotency by serum induction (Toma et al., 2001). The differentiation potential of the UKs (within four passages) from six adult donors was firstly assayed by culturing these cells with DMEM-F12 supplemented with different FBS concentration (10, 5, or 1%). However, most cells floated and only a few round cells reattached back to the surface of rapidly differentiated keratinocytes and formed an “island”-like cell cluster after 20 h. Within 3 weeks, these round cells differentiated into typical columnar epidermal cells, without other specific cell types being discerned (Fig. 2).

Mesodermal cell types and neural cells were not generated from UKs by nonacclimatized serum induction. (
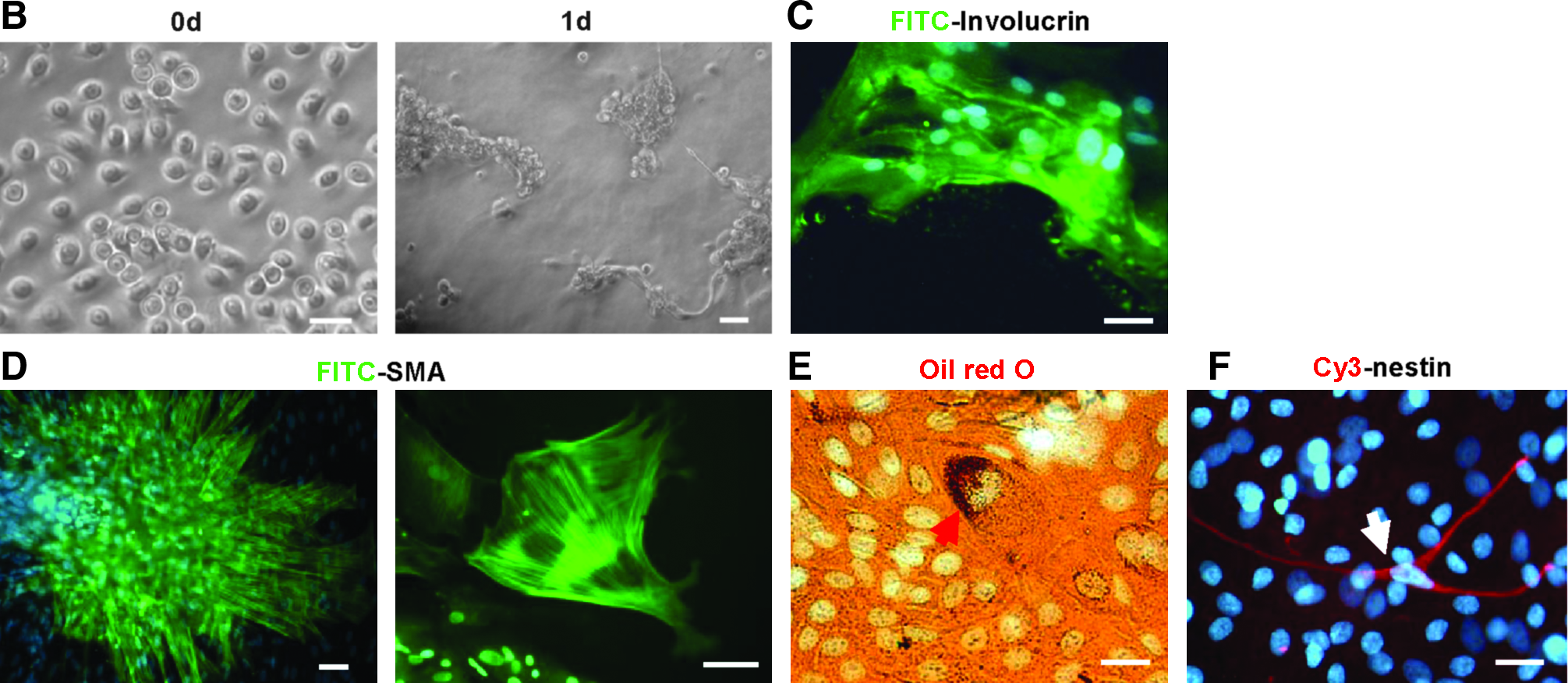
Epilife can effectively maintain the undifferentiated state of UKs. So, we postulated that the multipotency of these cells might be awakened if the preferential epidermis differentiation could be effectively prevented at the initial and early induction stage by the appropriate blending of EpiLife and the induction medium. Namely, UKs can be cultured by gradually raising the amount of the serum concentration, meanwhile gradually diminishing the amount of Epilife culture medium. We termed this approach “acclimatized serum induction.” To validate this hypothesis, we tested the initial volume ratio of Epilife and the serum differentiation medium and observed that it was better for multipotent induction of the UKs when these cells were initially cultured with a 20% proportion of 5% FBS-containing DMEM-F12 medium and subsequently treated as illustrated in Figure 3A. Similarly, island-like cell aggregates were formed after 1-day culture (Fig. 3B). Although most UKs formed terminally differentiated epidermal cells after 2 weeks (Fig. 3C
Adult human UKs generated mesodermal cell types and neural cells by acclimatized serum induction. (
Committed acclimatization induces UKs generating desired cell types
Most keratinocytes cannot survive in 100% lineage-committed culture medium, and the relic cells rapidly differentiate into epidermal cells within 1 week (data not shown). Acclimatized serum induction effectively hampered the preferential differentiation of UKs into epidermal cells and promoted the generation of multilineage cell types. So, we adopted this strategy to determine whether these cells could be differentiated to the desired cell types by lineage-committed induction.
Adipogenic differentiation was induced by cells with isobutyl–methylxanthine, dexamethasone, insulin, and indomethacin as described previously (Pittenger et al., 1999). The optimized acclimatization procedure for adipogenic differentiation of undifferentiated keratinocytes illustrated in Figure 4A could maximally block epidermal differentiation. After 1-week acclimatization, lipid-rich vacuoles within cells were discerned; the lipid vacuoles continued to develop over time, coalesced, and eventually filled the cells (Fig. 4B). After 3 weeks, more than 15 % of the cells were committed to adipocytes, which expressed peroxisome proliferation-activated receptor γ2 (PPARγ2), lipoprotein lipase (LPL), and the CCAAT/enhancer binding protein α (C/EBPα) (Fig. 4D).
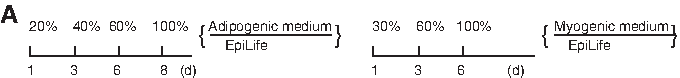
Desired mesodermal cell lineages generated from UKs by committed acclimatization induction. (


We used supernatant of cultured rat myocardial cells and retinoic acid (RA, 0.5 μM) to promote myogenic differentiation of UKs. We found a balanced acclimatization modality illustrated in Figure 4A for blocking epidermis formation and promoting myogenic efficiency and velocity. A large number of SMA-positive cells (10–65%) with the morphology of smooth muscle cells or myofibroblasts were observed after 1-week treatment, and muscle cells with double nuclei were detected after 3 weeks (Fig. 4C). RT-PCR analysis demonstrated that these cells expressed MyoD, Myf5, and sm22α (Fig. 4D).
For neurogenic differentiation of UKs, cells were first stimulated by adding 0.5 mM β-mercaptoethanol, 45 ng/mL basic fibroblast growth factor, 20 ng/mL epidermal growth factor, 0.5 mM RA, 0.5 mM VPA, 2000 U/mL LIF, 100 mM L-glutamine, 30 mM glucose, 5 μg/mL insulin, 1 mM sodium pyruvate, and 2% DMEM-F12 (containing 2% FBS) into EpiLife medium for 36 h. Cells were then cultured for 2 days in medium with 20% DMEM-F12 (containing 2% FBS) and 80% EpiLife containing 20 ng/mL bFGF, 20 ng/mL EGF, 0.5 μM RA, 1 × B27, and 100 μg/mL BHA. Finally, the amount of DMEM-F12 (containing 2% FBS) was increased to 100% at a 20% increase every 2 days. This treatment induced about 30% of round keratinocytes producing small spinous processes after 2-day stimulation (Fig. 5A). After 10 days, terminally differentiated epidermis began to appear. At 2 weeks, we observed nestin-positive cells (3–15%) (Fig. 5B). After 3 weeks, 2–11% cells expressed neuron-specific β3-tubulin, and 1–3% of cells expressed the glial marker GFAP (Fig. 5C and D).
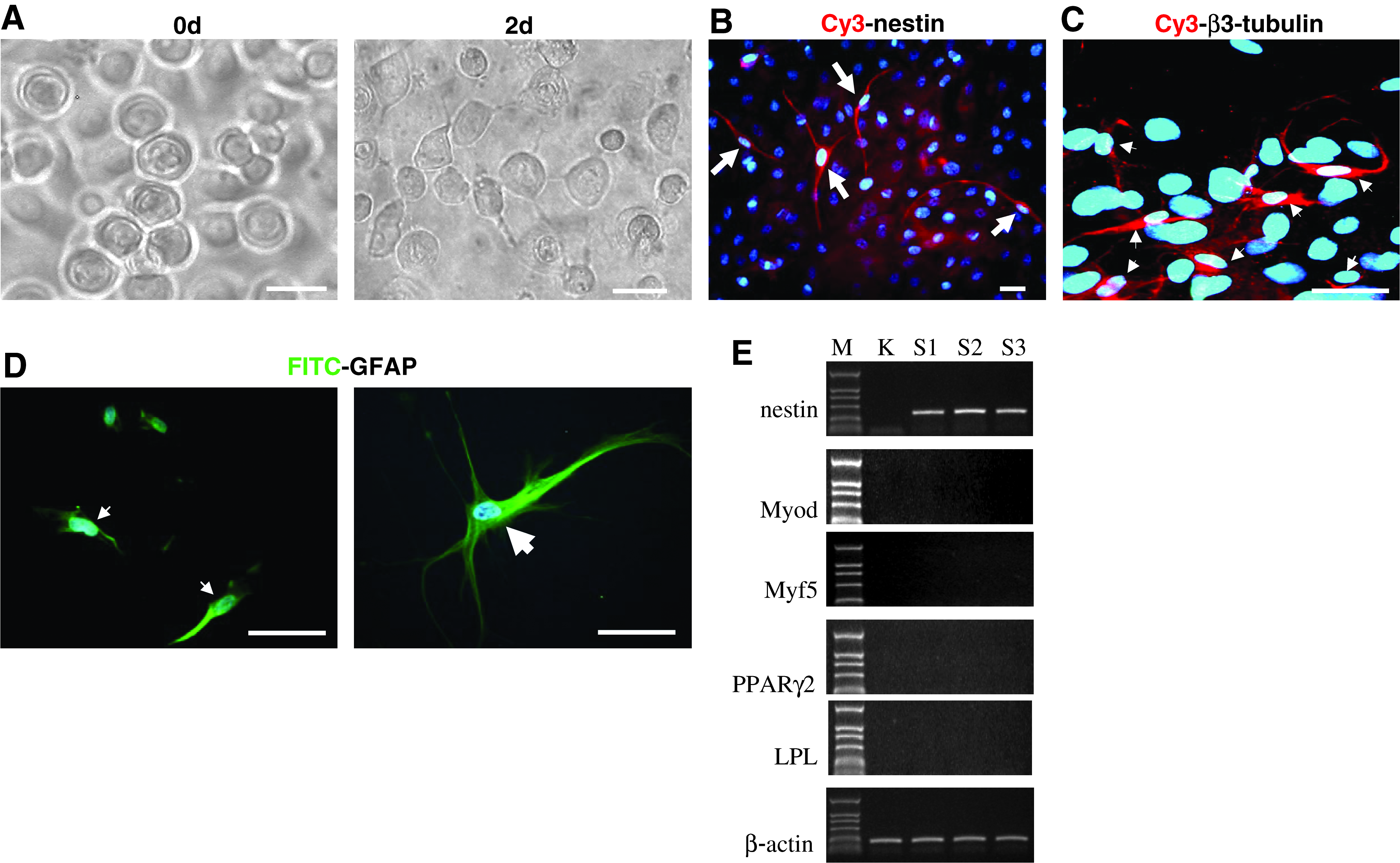
Neural cell lineage generated from UKs by committed acclimatization induction. (
Unlike the nonspecific acclimatization induction by serum, the entire induction of committed acclimatization appeared to be solely responsible for the controlled generation of the desired cell lineage, except for terminally differentiated epidermal cells, on the basis of phenotypic characterization and RT-RCR analysis (Fig. 4D and Fig. 5E).
In this study, at least 15 samples at different passages (most within three passages) were performed for lineage-committed acclimatization induction and the similar results were observed. The induction rate appeared to be positively correlated with the proportion of cells positive for CK14, a well-documented marker of UKs (Blanpain and Fuchs, 2009). As shown in Figure 6A, more than 60% cells can be labeled with CK14 by immunofluorescence analysis within passage 1. The CK14-positive cells gradually decreased along with cell passage. Meanwhile, the percentages of SMA-, Oil red O-, and nestin-positive cells generated by acclimatized UKs were also gradually decreased along with the decrease of CK14-positive cells (Fig. 6B). In addition, we attempted to induce differentiation of keratinocytes at more late passages (greater than passage 5) from three samples by lineage-committed acclimatization. However, except for sporadic SMA-positive cells (<1%), Oil red O- and nestin-positive cells cannot be easily detected after 3-week induction (data not shown). These results suggested that adult UKs only at early passages (within passage 5) could be easily induced into other lineages.
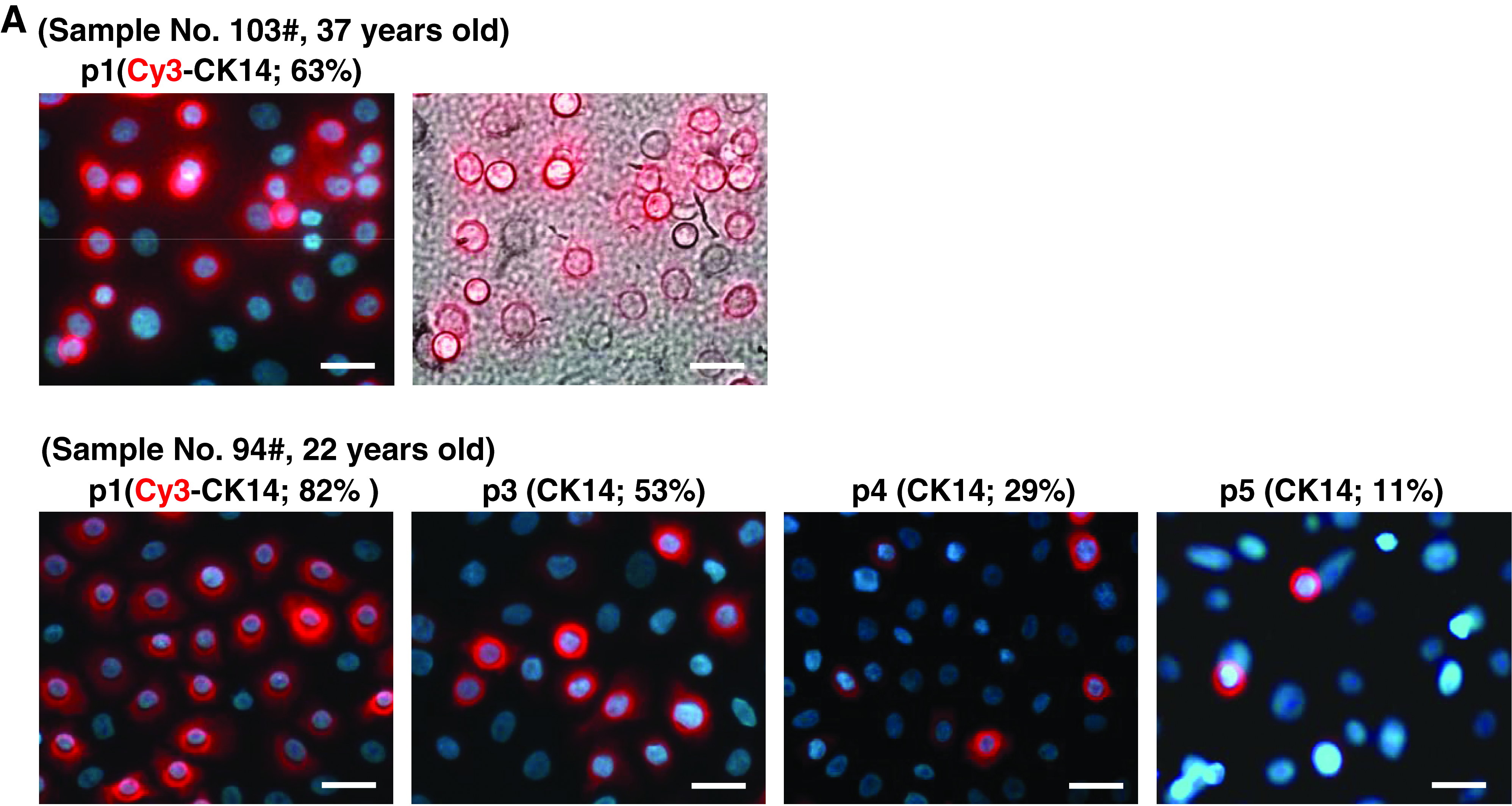
The multipotency of keratinocytes isolated from basal layer of adult human foreskin epidermis positively associated with level of CK14-positive cells. (

Discussion
Accumulated evidence supported the notion that if tissue-specific stem cells are exposed to appropriate foreign environments, as occurs in vitro or following heterotopic transplantation, they can express a wider differentiation potential (Banas et al., 2007; Jahagirdar and Verfaillie, 2005; Wagers and Weissman, 2004). However, UKs derived from the basal layer of epidermis seem to be unipotent precursors that only differentiate into epidermal cells (Fuchs, 2007). In the present study, we developed an acclimatization strategy, by which UKs at early passages (within passage 5) were revealed to be multipotent. UKs isolated from the basal layer of adult human foreskin can differentiate into other multiple lineages including adopcytes, myocytes, and neurocytes by noncommitted serum acclimatization induction. By committed acclimatization induction, UKs can differentiate exclusively into the adipocytic, myogenic or neurogenic lineages. These findings first reveal the multipotency of adult human UKs and underscore that UKs may become therapeutically promising cells in regenerative medicine.
There are many different types of stem cells in the skin, including melanoblast stem cells, mesenchymal stem cells, neural-like stem cells, hair follicle stem cells, sebaceous gland stem cells, and interfollicle epidermal stem cells. Although some of them such as melanoblast stem cells, mesenchymal stem cells, and neural-like stem cells have been realized to be multipotent (Crigler et al., 2007; Motohashi et al., 2009; Toma et al., 2001, 2005), they are difficult to be isolated and require long-term culture to expand to a large amount. These are obvious limiting factors for their application in regenerative medicine. Interfollicle stem cells are enriched in keratinocytes present in the basal layer of epidermis (Blanpain and Fuchs, 2009; Fuches, 2007; Fuchs and Horsley, 2009). Compared with other types of stem cells in skin, the cellular population of keratinocytes from the basal layer of epidermis is readily accessible and easily cultured and expanded in vitro. Since the 1970s, with the publication of the work by Rheinwald and Green (1975), serial culture of human keratinocytes became possible (Gragnani et al., 2008; Li et al., 1998; Liang and Bickenbach, 2002; Papini et al., 2003). The cultured basal epidermal keratinocytes has already been realized to be easily expanded to a therapeutic amount for treating burns patients as early as in 1990s (Green, 1991). Human foreskin does not contain hair follicles. In this study, adult human keratinocytes were isolated from the basal layer of foreskin in more than 100 donors and cultured in Epilife medium. This culture allows the large proliferation of UKs and effectively blocks their differentiation (Fig. 1); however, after the third passage, CK14-positive cells were markedly decreased (Fig. 6A), demonstrating this culture method is suitable for short-term culture of UKs. In addition, human keratinocytes cultured in this system maintain stable karyotyping (Fig. 1D) and appear to be free of contaminating melanocytes or dermal fibroblasts (Fig. 1C). Because UKs are easily prepared and expanded, they may have obvious advantages over other stem cells as a cell source for regenerative medicine if they were multipotent.
Different with “pure” differentiation induction used in other stem cells such as MSCs, “pure” noncommitted serum induction of UKs leads to a full epidermal differentiation (Fig. 2), and most UKs can not survive in “pure” lineage-committed induction medium. However, acclimatization induction provokes the multipotent nature of UKs (Figs. 3–5). Similar to the conditional induction of skin-derived other stem cells and other tissue stem cells into multipotency (Crigler et al., 2007; Motohashi et al., 2009; Toma et al., 2005), our acclimatized induction of UKs into other cell lineages further demonstrated that the preferential selection of tissue-specific stem cells into the same lineage as their origin can be disrupted by the specific external environment. Therefore, acclimatization induction is a simple concept and also helps us resolve a long-term puzzle of some magnitude. Although the differentiation of UKs to other cell types is only testified by immunostaining in this study and further functional assays are being investigated in our laboratory, our acclimatized induction strategy reveals that differentiation of basal layer cells from human epidermis into epidermal cells is not the unique selection as was previously observed.
Optimal culture can induce stem cells to transdifferentiate into desired cell types, which is believed to be caused by the specific alteration of transcriptome-wide “noise” of cells under certain culture conditions (Hoffmann et al., 2008). Alteration of the transcriptional “noise” is proposed to mainly control the lineage choice of mammalian progenitors (Chang et al., 2008). The term “acclimatization induction” represents a gradual environment change, which can peaceably change the habitus or differentiation fate of an organism and cellular phenotype (Hawes et al., 2007; Mus et al., 2007; Savage and Schmidt, 2008), and, even more, alter the transcriptome-wide “noise” of stem cells (Hoffmann et al., 2008). “Pure” differentiation induction leads UKs rapidly differentiating into its original lineage or to death, suggesting that “pure” induction is not beneficial to maintaining the persistent undifferentiated status of UKs. Eplife medium can effectively block epidermization of UKs and maintain their undifferentiated status at least at early passages (within passage 5). Our study indicates that only acclimatization induction can provoke the multipotent nature of UKs. A reasonable explanation is that only in the persistent undifferentiated status (in the presence of Epilife medium), UKs can be pushed to a transcriptional “noise” status by gradually increasing induction medium, where the differentiation of UKs to other lineages can be initiated. Notably, only partial UKs were differentiated into other cell lineages even by acclimatized induction, further suggesting that the specific alteration of transcriptome-wide “noise” is a stochastic event in cell pools (Kaern et al., 2005; Losick and Desplan, 2008).
An implication of the findings present here is a therapeutic one. Cell-replacement therapies show particular promise in the nervous system, where transplanted either neural or embryonic stem cells can promote functional recovery in injured nervous system (Coutts and Keirstead, 2008; Okano et al., 2003; Tator, 2006). However, although the therapeutic potential of both neural and embryonic stem cells is clear, a number of important problems remain (Barnabé-Heider and Frisén, 2008; Bjorklund and Lindvall, 2000). Neural stem cells are not easily accessible and a major caveat to the use of ES-derived cells is the well-recognized risk of teratoma formation after transplantation. Fetal tissue is the current tissue source for human neural and stem cells, raising important ethical issues and involving heterologous transplantation. In this regard, the fact that UKs are generated from a potentially autologous, accessible adult tissue source, epidermis, and that they can readily generate neural cell types may provides a potential solution to these problems. In fact, it was observed, as early as in 1990s, that the function of nerve fibers in patients with skin defects can be improved by keratinocyte transplantation (Tóth Kása et al., 1994). In addition, the fact that UKs can simultaneously generate epidermal cells, muscle, and neural cells by a noncommitted acclimatization induction suggests that UKs might provide a potential cell source for treating combined trauma disease including skin, muscle, and nerve.
Together, we first demonstrated the multipotency of UKs isolated from the basal layer of adult human epidermis. Acclimatization was required to tame these stem cells showing their plasticity. Other selective differentiation should provide further understanding of keratinocyte stem cells. These cells may provide an alternative to using embryonic stem cells or other somatic stem cells that are more difficult to retrieve as therapeutic treatment for disease. Our data also suggested that establishing a suitable acclimatized microenvironment for these cells in vivo may provide a selectable strategy for wound healing in mammals and perfect regeneration of damaged tissues (Davenport, 2005).
Footnotes
Acknowledgments
This work was supported by funds from the Ministry of Science and Technology of China (2005CB522603 and 2010CB912802) and the National Natural Science Foundation of China (30870507, 30730090, 30670966).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.