
Abstract
Abstract
Embryonic stem (ES) cells constitute a very important tool for regenerative medicine today. These ES cells, and human ES cells in particular, are almost all derived from embryos obtained by in vitro fertilization (IVF) and from in vitro culture (IVC); however, such in vitro manipulated embryos often show abnormal genomic imprinting, which can lead to the development of various diseases. Nevertheless, several reports have evaluated ES cells derived from in vitro manipulated embryos. In this study, we established ES cells derived from both in vivo and in vitro developed blastocysts (Vivo ES cells and Vitro ES cells, respectively) to compare the methylation status of imprinted genes and gene expression patterns. At very early passages, Vitro ES cells showed an increase in abnormal genomic imprinting compared to Vivo ES cells. In addition, we found that the gene expression patterns of several methylation related-genes frequently shifted to promote demethylation and to inhibit methylation in early-passage Vitro ES cells. In contrast, at later passages, we found no significant differences between Vivo and Vitro ES cells. In conclusion, it is advisable to use early passage Vivo ES cells whenever feasible, or to select ES cell lines with a normal epigenotype.
Introduction
Genomic imprinting is the preferential silencing of one parental allele by epigenetic DNA methylation. For example, the expression levels of the H19 imprinted gene are regulated by an upstream differentially methylated region (DMR) (Bartolomei et al., 1991; Ferguson-Smith et al., 1991; Pfeifer, 2000). The H19 mRNA is transcribed from the unmethylated maternal allele, but is not transcribed from the methylated paternal allele. In mice, the erasure of genomic imprinting takes place in germ lines between embryonic day (E) 10.5 and E12.5, around the time when primordial germ cells enter the gonads (Szabo and Mann, 1995). Following erasure, DNA methylation patterns are then reestablished in a sex- and sequence-specific manner during gametogenesis. These methylation imprints are then stably maintained during cellular division and differentiation in somatic lineages. The expression levels of imprinted genes are regulated not only by DNA methylation but also by histone modification (Fournier et al., 2002); however, DNA methylation, including genomic imprinting, is regarded as more stable than histone modification in preimplantation embryos (Kim and Ogura, 2009) and brain (Yamasaki et al., 2005).
For the establishment and maintenance of DNA methylation, various enzymes and proteins are indispensable; for example, the cytosine-guanine (CpG) DNA methyltransferases, Dnmt1, Dnmt3a, and Dnmt3b, which coordinately regulate CpG methylation in the genome (Li et al., 1992; Okano et al., 1998, 1999). Dnmt1 is involved in maintenance activity, whereas Dnmt3a and Dnmt3b are mainly responsible for the creation of new methylation patterns. Stella (PGC7), a primordial germ cell and embryonic stem (ES) cell marker, protects against DNA demethylation in early embryogenesis (Nakamura et al., 2007). Zfp57, a putative KRAB zinc-finger protein, is also required for the postfertilization maintenance of maternal and paternal methylation imprints at multiple imprinted domains (Li et al., 2008). In contrast, the existence of active demethylation factors has rarely been reported in mammals, although the finding that Gadd45 (growth arrest and DNA damage inducible protein 45) interacts with the nucleotide excision repair (NER) complex and promotes DNA demethylation suggests that Gadd45 recruits the DNA repair machinery to specific sites to replace methylated cytosines with unmethylated ones (Barreto et al., 2007; Ma et al., 2009).
The culture conditions of fertilized embryos can also influence the methylation status of genomic imprinting. For example, a suboptimal culture medium can cause aberrant genomic imprinting of the H19 gene, whereas embryos cultured in potassium simplex optimized medium with added amino acids (KSOMAA) show global gene expression, genomic imprinting, and embryo development resembling that found in in vivo developed embryos (Doherty et al., 2000). Long-term culture of ES cells also affects the methylation status of imprinted genes and their totipotency (Dean et al., 1998). It is unclear what causes DNA methylation instabilities of imprinted genes, although some of the factors described above may be related to the instability of imprinted genes.
Mouse ES cells (Evans and Kaufman, 1981; Martin, 1981) can give rise to ectodermal, endodermal, and mesodermal germ layers both in vivo and in vitro. In human ES cells, several studies have recently provided evidence of the efficient induction of endoderm, mesoderm, and ectoderm, and many of their downstream derivatives (Murry and Keller, 2008), and these reports offer broad perspectives for regenerative medicine. However, all human ES cell lines are established from in vitro manipulated embryos that often show abnormal genomic imprinting, which can lead to an increase in the frequency of diseases. Therefore, although it is very important to compare the epigenetic composition of Vivo and Vitro ES cells, there have been few reports characterizing their differences. In this study, we have compared the methylation status of imprinted genes and the gene expression patterns of both Vivo and Vitro ES cell lines in mice, and we have concluded that Vitro ES cells are similar to Vivo ES cells, except at very early passages.
Materials and Methods
Mice
The C57BL/6J mouse strain (B6) was purchased from Charles River Japan. The PWK mouse strain (RBRC00213) was provided by RIKEN BRC (Japan), which is participating in the National Bio-Resource Project of the MEXT, Japan. All animal experiments were conducted according to the guidelines of the Animal Care and Experimentation Committee, Gunma University, Showa Campus, Japan.
Embryo collection and culture
B6 females were superovulated by the injection of five units of pregnant mares' serum (PMSG; ASKA Pharmaceutical, Tokyo, Japan) and 48 h later by five units of human chorionic gonadotrophin (hCG; ASKA Pharmaceutical). For the production of in vitro cultured embryos, cumulus–oocyte complexes were collected in TYH medium (Toyoda et al., 1971), from oviducts 16 to 17 h post-hCG injection (p.i.). Spermatozoa were collected from the caudal epididymis of adult PWK, and B6 males and were capacitated by preincubation for 1 h in TYH medium. Cumulus–oocyte complexes were inseminated with capacitated spermatozoa in a humidified atmosphere of 5% CO2, 95% air at 37°C. Six hours after insemination, the B6xPWK F1 (BPF1) or B6xB6 (B6) fertilized oocytes were washed and cultured in M16 medium supplemented with 0.1 mM EDTA/2Na. The embryos developed into blastocysts at 4.5 days p.i (in vitro blastocyst). In contrast, embryos allowed to develop in vivo (in vivo blastocyst) were collected from females killed at 3.5 days p.i. by flushing their uteri with M2 medium.
Generation of ES cell lines
Both in vitro blastocysts at 4.5 days p.i. and in vivo blastocysts at 3.5 days p.i. were transferred onto gelatinized tissue culture wells (two to three blastocysts per well of a four-well multiplate) and cultured for 7 days in ES medium, DMEM (Gibco, Gland Island, NY, USA) containing 17.5% Knockout SR (Gibco), following standard procedures (Horii et al., 2008; Robertson, 1987). After 7 days, inner cell mass (ICM) outgrowths were harvested in 0.25% trypsin-EDTA (Gibco), disaggregated by mouth pipetting, and plated onto gelatinized tissue culture wells in ES medium (passage 1). Clones resembling ES cells morphologically were picked and disaggregated a second time. They were then expanded and passaged prior to use.
Alkaline phosphatase (AP) staining
Histochemical staining for AP activity was performed as previously described (Buehr and McLaren, 1993).
Generation of embryoid bodies
For embryoid body (EB) formation, ES cells were detached and dissociated into single cells with trypsin-EDTA and then plated onto a bacteriological dish in 10 mL of DMEM supplemented with 15% fetal bovine serum (Hyclone, Logan, UT, USA), nonessential amino acids (0.1 mM), and 2-mercaptoethanol (0.1 mM).
RNA isolation and reverse transcription
Total RNA was purified from ES cells and EBs using TRIZOL reagent (Invitrogen, Carlsbad, CA), following DNaseI treatment. One microgram of purified RNA was reverse transcribed using Superscript II (Takara Bio, Otsu, Japan) and Oligo(dT)12–18 primer (Takara Bio) in a total volume of 20 μL. For quantitative real-time polymerase chain reaction (PCR), cDNA was diluted 1:10 in distilled water.
Quantitative real-time PCR
Quantitative real-time RT-PCR was performed using SYBR Premix Ex Taq (Perfect Real Time; Takara Bio). The PCR mixture consisted of 2 × SYBR Premix Ex Taq, 10 μM forward and reverse primers, 50 × ROX reference dye, and 2 μL of template cDNA in a total volume of 12.5 μL. The cocktail was activated by heating initially at 95°C for 10 sec. The subsequent PCR reaction was carried out at 95°C for 5 sec and at 60°C for 30 sec for 40 cycles in an ABI 7700 Sequence Detector. In each run, the dilution series of cDNA from ES cells or EBs was amplified to serve as a standard curve for the calculation of relative quantities of the target gene using Sequence Detector Software v1.7 (comparative CT method). The Gapdh gene was used to standardize the data. All results were obtained from at least two independent experiments and each assay was duplicated. PCR amplification was performed using the primer sets shown in Table 1.
Allele-specific expression analysis
First-strand cDNA was subjected to PCR, which was carried out using LA Taq HS (Takara Bio) and the primer sets shown in Table 1. H19 gene polymorphisms were detected by RFLP (restriction fragment-length polymorphism). Following RT-PCR, H19 products were digested with Bgl I at 37°C for 3 h and separated on a 2% agarose gel in 0.5 × TAE.
DNA isolation and methylation analysis
DNA was isolated from each ES cell line. Bisulfite treatment was carried out using an Epitect Bisulfite kit (Qiagen, Hilden, Germany), according to the manufacturer's instructions. PCR amplification of H19 DMR, Snrpn DMR1, and Igf2r DMR2 was performed for each set of isolated cells using the primer set shown in Table 1. The amplification consisted of a total of 35 cycles at 94°C for 10 sec, 55°C (H19) or 60°C (Igf2r and Snrpn) for 30 sec, and 72°C for 60 sec in a GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA, USA). PCR products were subcloned into the TA cloning vector (pCR 2.1; Invitrogen). Positive clones in each sample were sequenced using the Big Dye terminator method (ABI PRISM 3100; Applied Biosystems). Parental alleles were determined by SNPs (Table 2). Combined bisulfite restriction analysis (COBRA
Immunoblot analysis
Whole cell extracts were subjected to 10% SDS-PAGE gel electrophoresis and the protein were transferred to a PVDF membrane. The following antibodies (1:1,000) were used for immunoblotting: anti-Oct3/4 (sc-5279, Santa Cruz, CA, USA), anti-Dnmt3b (kindly donated by Dr. S. Tajima) and anti-α-tubulin (CP06, Calbiochem, Darmstadt, Germany). Signals were detected by chemiluminescence (ECL Plus, GE healthcare, Buckinghamshire, UK) on a CCD camera (LAS-3000, Fujifilm, Tokyo, Japan). Protein band intensities were normalized to those of α-tubulin using Image J software (NIH).
Statistical analysis
Data are shown as means and standard deviations. The Student's t-test was used for methylation, gene expression, and immunoblot analyses, and a p-value of <0.05 was considered significant. The statistical analyses were performed with Excel X and Statcel2 for Macintosh. QUMA (QUantification tool for Methylation Analysis; http://quma.cdb.riken.jp/) was used for statistical analyses of the bisulfite sequencing of CpG methylation (Kumaki et al., 2008).
Results
Establishment of ES cell lines
Freshly collected in vivo blastocysts and in vitro cultured blastocysts were prepared for the establishment of ES cell lines. The embryos obtained in vivo had developed to the blastocyst stage at the time they were flushed (3.5 days p.i.), whereas 1 extra day in culture (4.5 days p.i.) was necessary to obtain in vitro cultured blastocysts (Fig. 1A). Both in vivo and in vitro-derived blastocysts were used for establishing ES cell lines. All ES cell lines obtained formed densely packed colonies and showed AP activity (Fig. 1B). The efficiency of ES cell establishment was not significantly different between in vivo and in vitro developed blastocysts (Table 3).
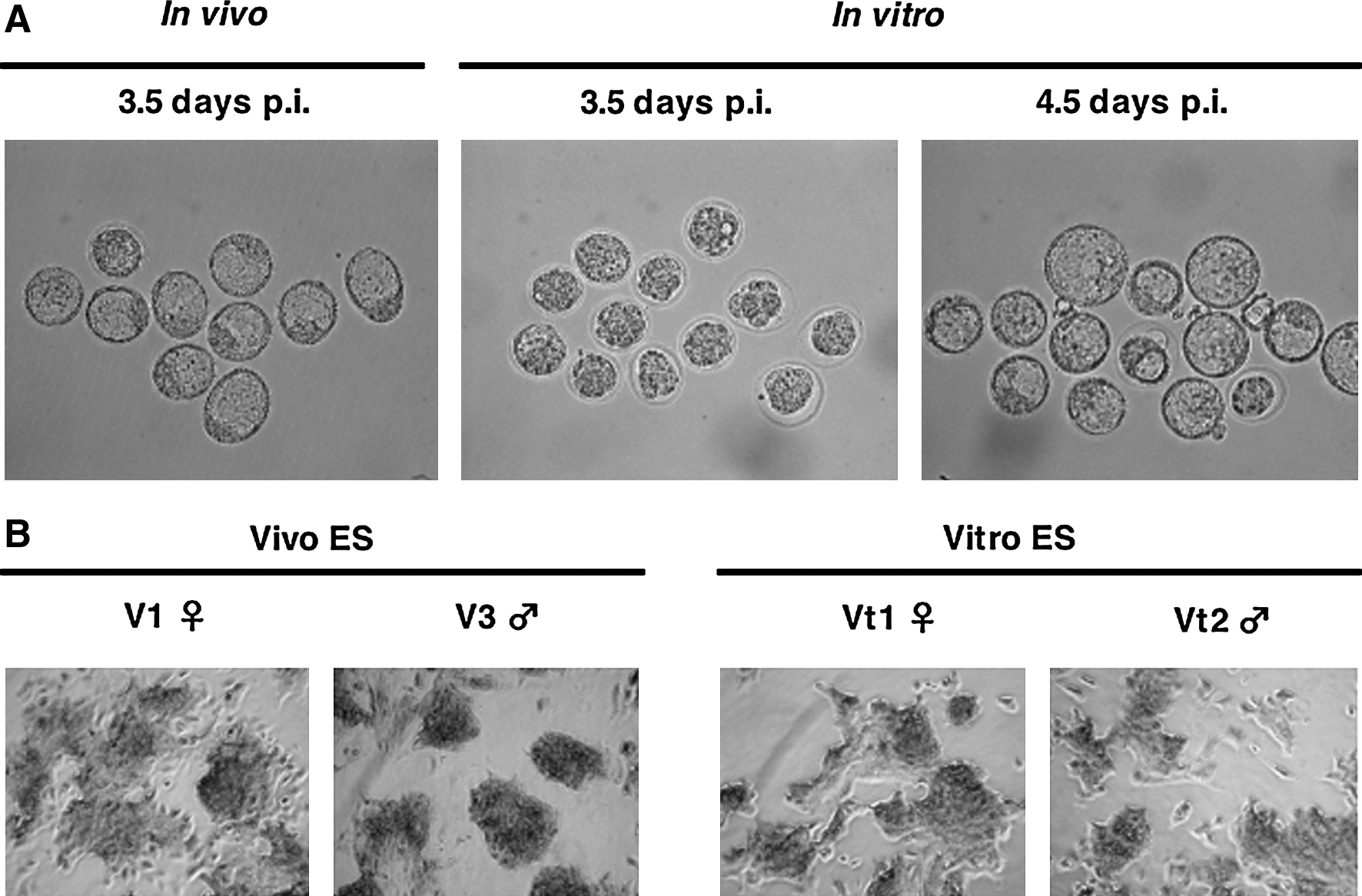
Establishment of Vivo and Vitro ES cells (BPF1 mouse strain). (
Methylation status and gene expression at early passages
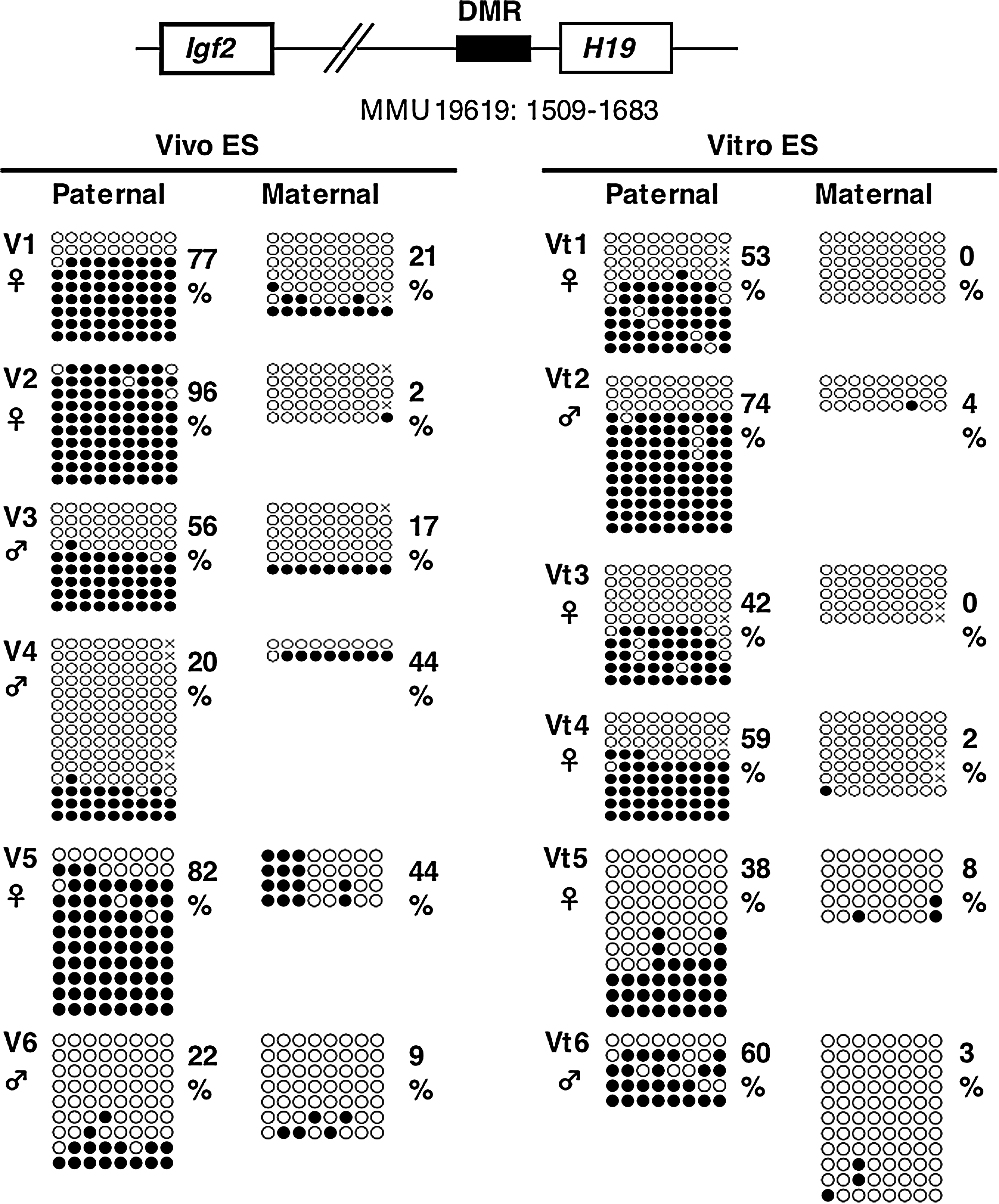
The imprinting of the H19 gene, one of the paternally methylated genes, is stably maintained during cellular division and differentiation, and is transcribed exclusively from the maternal allele. It was also thought that the imprint was maintained during preimplantation development (Pfeifer, 2000). In contrast, some researchers have demonstrated that normal imprinting can be disrupted during preimplantation development, resulting in biallelic expression of the H19 gene (Doherty, et al., 2000; Mann, et al., 2004; Sasaki, et al., 1995). To investigate whether Vitro ES cells take on abnormal imprinting from in vitro cultured blastocysts, we performed methylation analysis of the H19 DMR for early passage cells (passage 2). The results of bisulfite sequencing showed that the H19 DMR on the paternal allele was more widely demethylated in Vitro ES cells than in Vivo ES cells (Fig. 2), as was reported for in vitro cultured blastocysts (Doherty et al., 2000). Other imprinted genes, Snrpn DMR1, and Igf2r DMR2, did not show large differences (Fig. 3), although the average methylation levels of Vivo ES cells were higher than those of Vitro ES cells. COBRA analysis confirmed that the H19 DMR was significantly demethylated in Vitro ES cells compared to Vivo ES cells at the Hinf I site (Fig. 4). Igf2r DMR2 also showed significant differences in COBRA (Fig. 4), but not in bisulfite sequencing. On the basis of the bisulfite sequencing and COBRA results, the methylation status of H19 DMR was judged more unstable than that of other imprinted genes and satellite repeats.
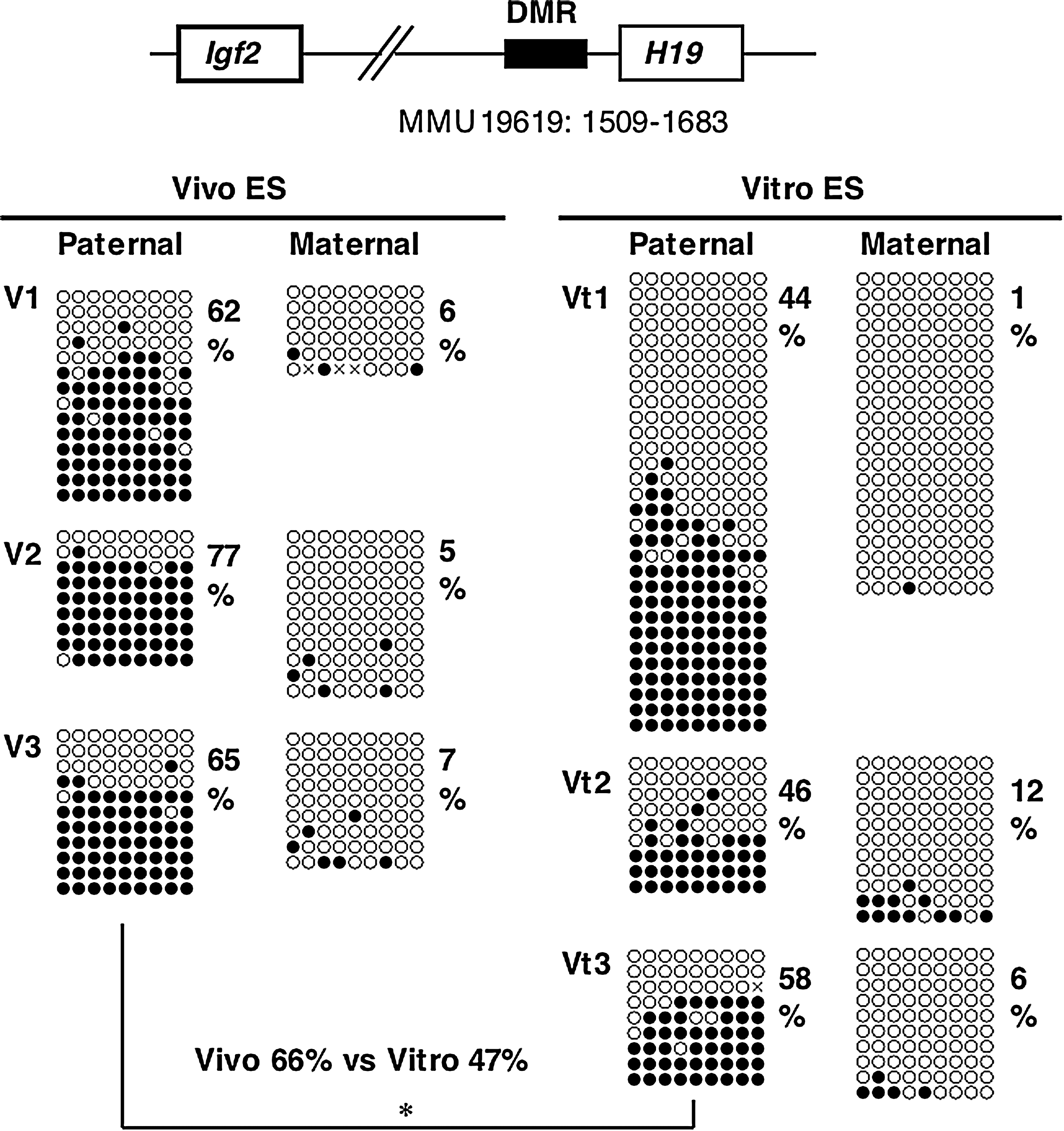
Methylation status of the H19 DMR at early passages (passage two) in Vivo and Vitro ES cells (BPF1 mouse strain). Bisulfite sequencing analysis of paternal and maternal alleles was carried out for Vivo and Vitro ES cells. The PCR product was subcloned, and clones were subjected to nucleotide sequencing analysis. The methylation status, either unmethylated (open circle) or methylated (closed circle), is indicated at each CpG site. Percentages of methylated CpGs are shown to the right of the sequences. Vitro ES cells show significantly greater demethylation than Vivo ES cells on the paternal allele of the H19 DMR (*p < 0.05).
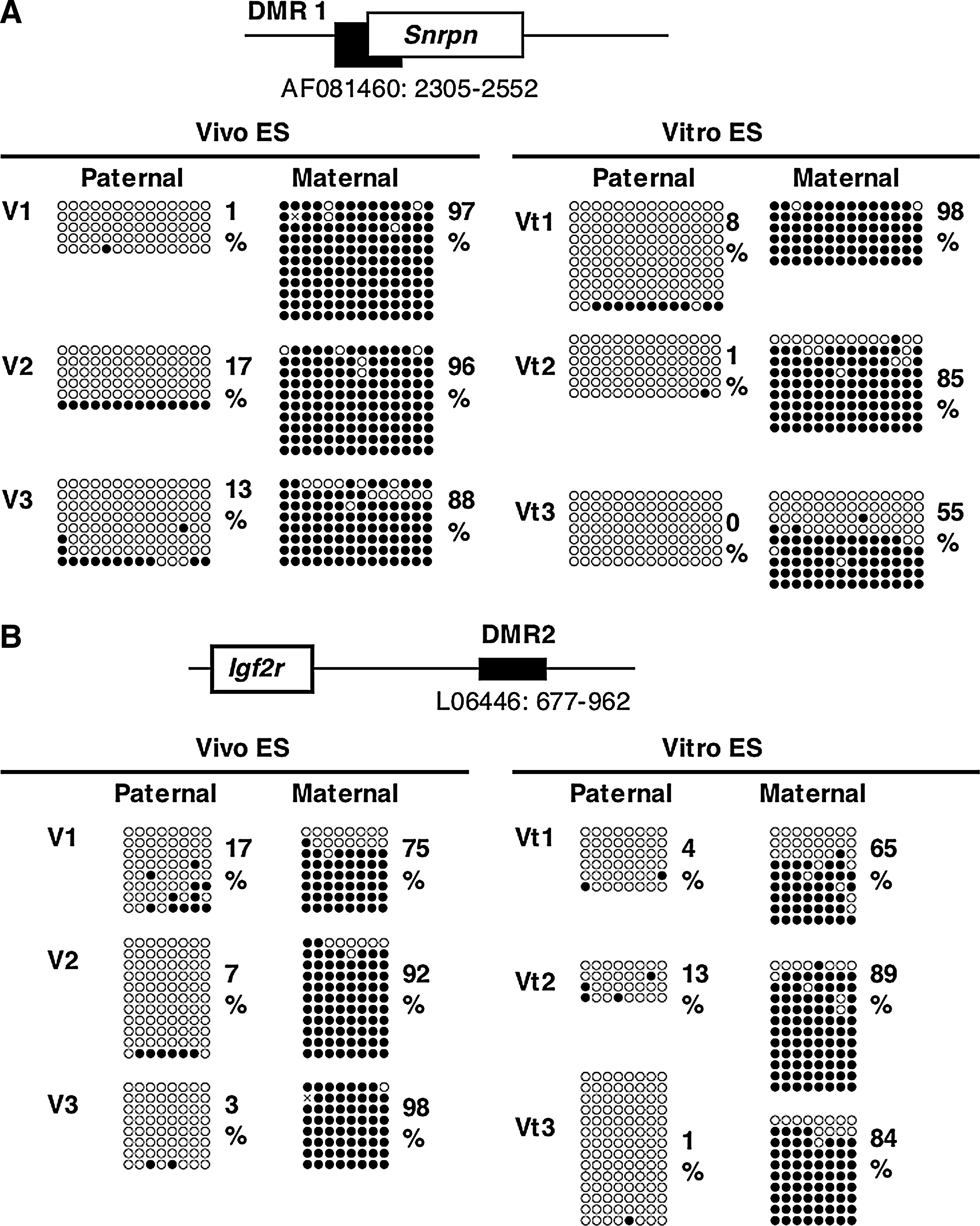
Methylation status of the Snrpn DMR1 and Igf2r DMR2 at early passages (passage 2) in Vivo and Vitro ES cells (BPF1 mouse strain). Bisulfite sequencing analysis of paternal and maternal alleles was carried out for Vivo and Vitro ES cells. The PCR product was subcloned, and clones were subjected to nucleotide sequencing analysis. The methylation status, either unmethylated (open circle) or methylated (closed circle), is indicated at each CpG site. Percentages of methylated CpGs are shown to the right of the sequences.
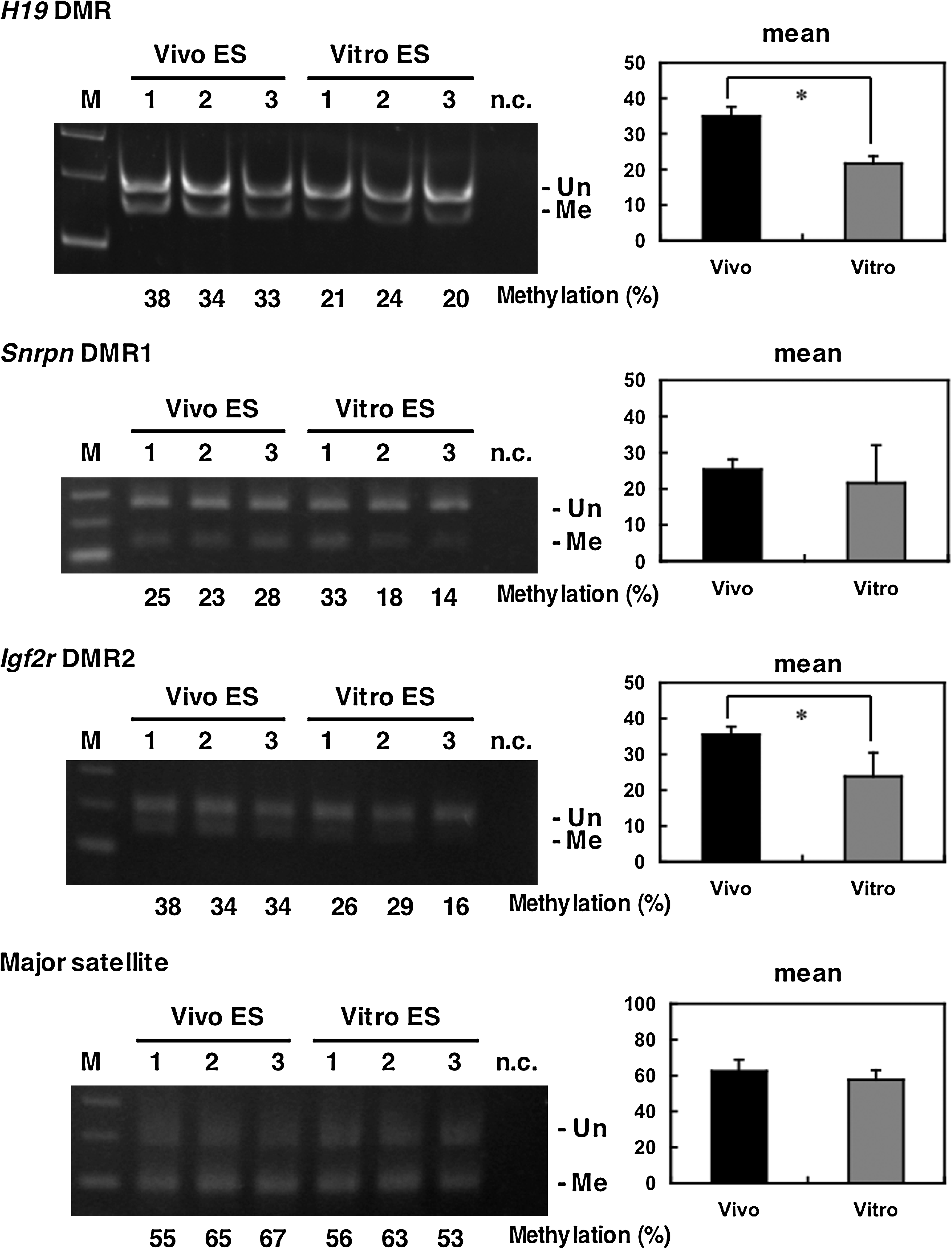
Methylation status of imprinted genes and satellite repeats at early passages (passage 2) in Vivo and Vitro ES cells (BPF1 mouse strain). COBRA analysis shows that Vitro ES cells are significantly demethylated in H19 and Igf2r DMRs (*p < 0.05). M: marker, n.c: negative control, Un: unmethylated, Me: methylated.
Next, we assessed gene expression patterns in ES cells at early passages by quantitative real-time RT-PCR. The expression of Oct3/4 mRNA, a pluripotent cell marker, was significantly higher in early passage Vivo ES cells than in Vitro ES cells; whereas other pluripotent marker genes showed no significant differences in expression levels between the two types of ES cells (Fig. 5A). Among the methylation-related genes, mRNA expression of the de novo DNA methyltransferase, Dnmt3b, was significantly higher in Vivo ES cells (Fig. 5B). Gadd45b mRNA expression, which is considered to be one of the putative demethylation factors, was higher in Vitro ES cells (Fig. 5B). On the other hand, there was no difference in Zfp57 and Stella mRNA expression, which is essential for the maintenance of genomic imprinting (Fig. 5A and B), although Zfp57 expression was significantly different in B6 ES cell lines (Supplemental Fig. 1; see online supplementary material at www.liebertonline.com). Thus, mRNA expression patterns of several methylation related-genes tend to shift, resulting in the promotion of demethylation and the inhibition of methylation in Vitro ES cells. Nevertheless, there were no significant differences in Oct3/4 and Dnmt3b expression as judged by immunoblot analysis (Fig. 5C).
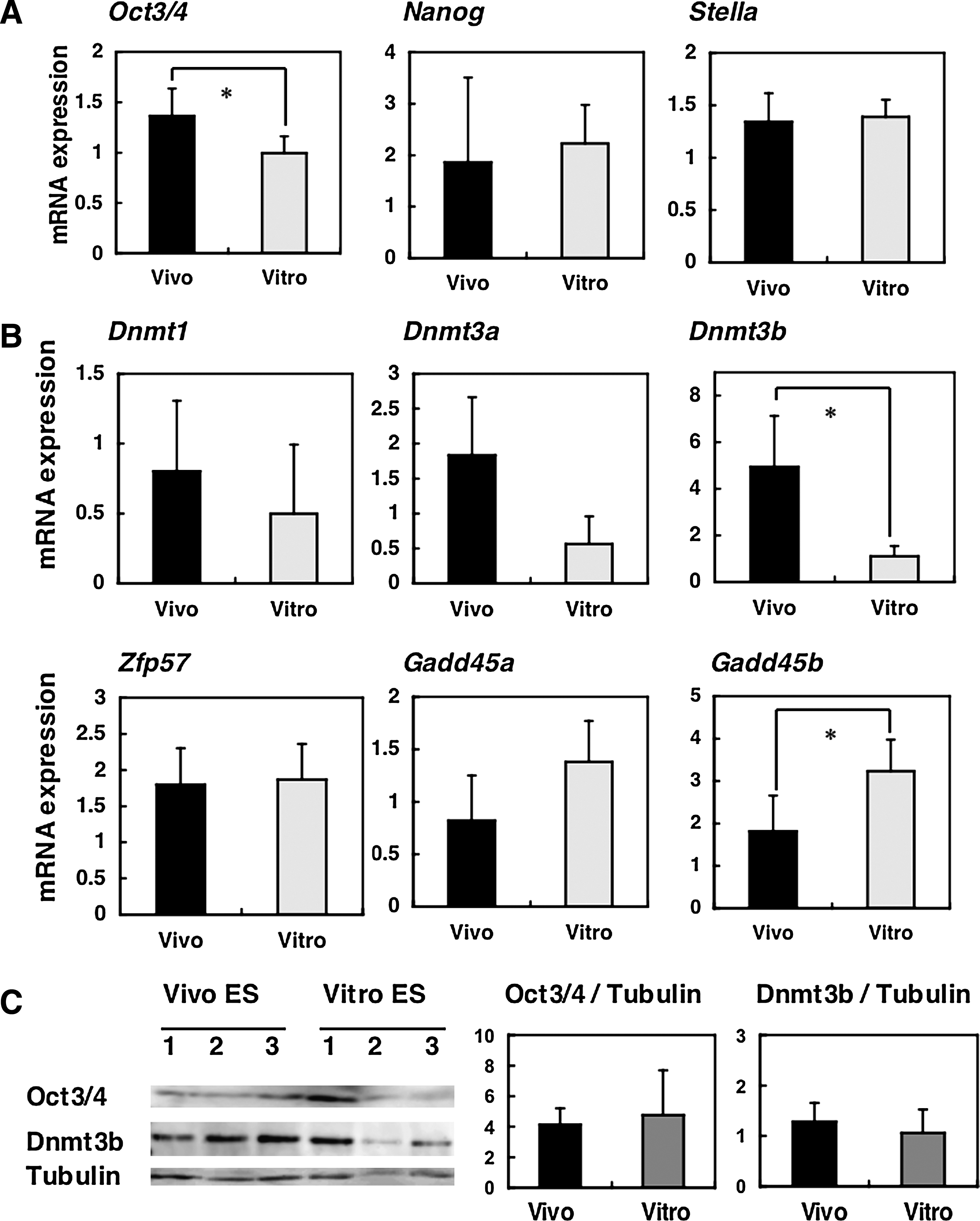
Expression patterns of early passage (passage 2) Vivo and Vitro ES cells (BPF1 mouse strain). (
Methylation status and gene expression at later passages
Both Vivo and Vitro ES cells were passaged several more times, and the methylation status of the H19 DMR was investigated at later passages. Results from COBRA analysis and bisulfite sequencing at passage five showed no significant differences between Vivo and Vitro ES cells (Figs. 6 and 7). Even Vivo ES cells exhibited highly demethylated alleles (V4 and V6 lines); in contrast, some Vitro ES cells had an almost normally methylated allele (Vt2 line). There was no difference between Vivo and Vitro ES cells in the methylation status of other genes (Fig. 7). This indicates that the methylation status of ES cells at later passages depends more on the character of individual cell lines than on the origin of the ES cells. We then assessed mRNA expression (Fig. 8A and B) and protein expression (Fig. 8C) by real-time RT-PCR and immunoblot analyses, respectively, at later passages and, as expected, we found no significant differences between Vivo and Vitro ES cells with respect to pluripotent marker genes and methylation-related genes.
Methylation status of the H19 DMR at later passages (passage 5) of Vivo and Vitro ES cells (BPF1). Bisulfite sequencing analysis for both paternal and maternal alleles was carried out for both Vivo and Vitro ES cells. The methylation status, either unmethylated (open circle) or methylated (closed circle), is indicated at each CpG site. Percentages of methylated CpGs are shown to the right of the sequences. There was no significant difference between the ES cell lines.
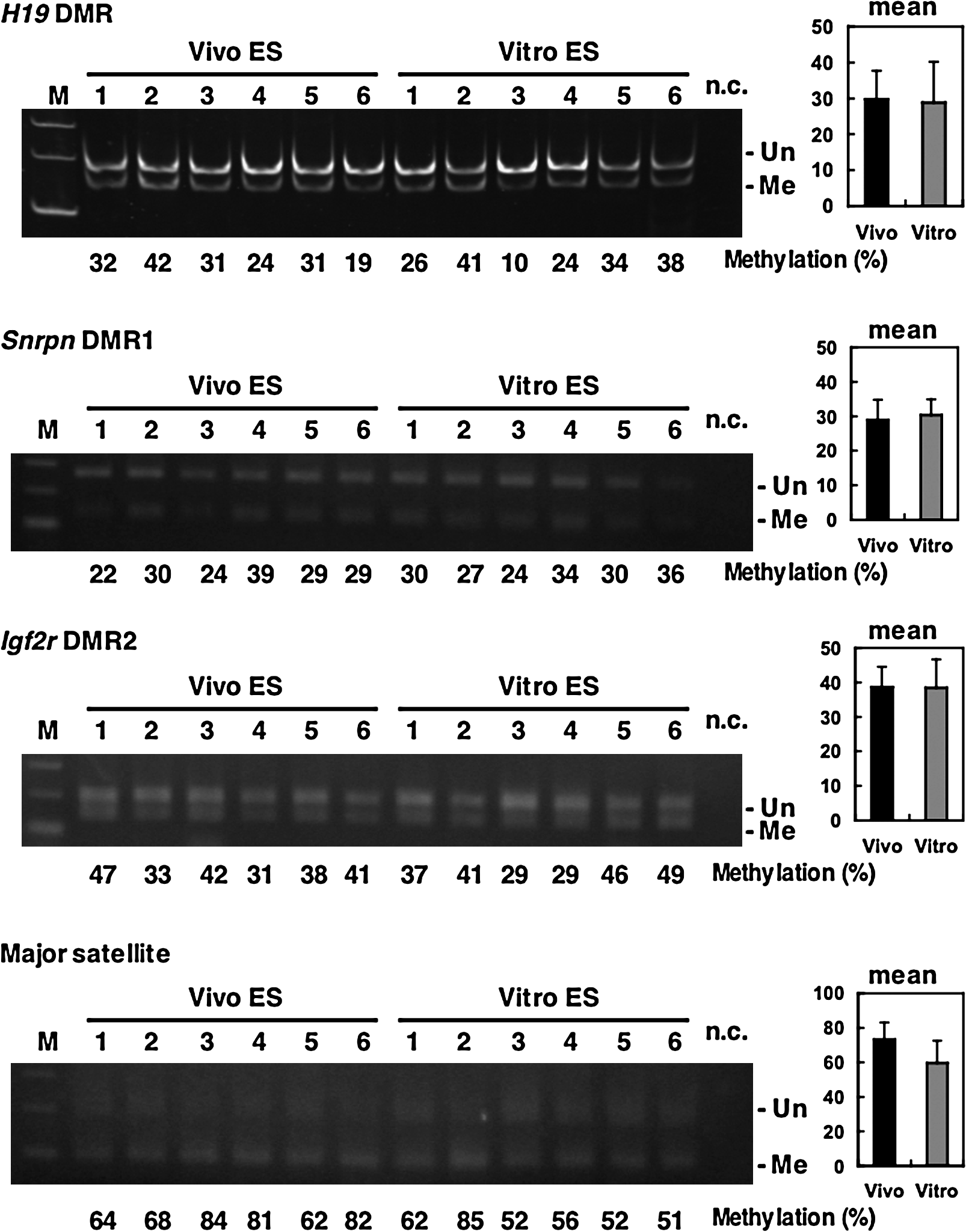
Methylation status of imprinted genes and satellite repeats at later passages (passage 5) in Vivo and Vitro ES cells (BPF1 mouse strain). There was no significant difference in the percentage of methylated sites for either ES cell line. M: marker, n.c: negative control, Un: unmethylated, Me: methylated.

Expression patterns of Vivo and Vitro ES cells (BPF1 mouse strain) at later passages (passage 6: V1–V4 and Vt1–Vt4, passage 5: V5, V6, Vt5, and Vt6). (
Gene expression after differentiation induction
The differentiation of ES cells in suspension into EBs mimics, at a rudimentary level, events occurring at the late pre- and early postimplantation stages (Doetschman et al., 1985; Martin, 1975; Stevens, 1975). We investigated the allelic expression of the H19 imprinted gene using EBs obtained at day 5 postsuspension culture. Although it is known that H19 RNA is transcribed from the unmethylated maternal allele, biallelic expression of H19 was frequently observed by PCR-RFLP analysis, regardless of the origin of the ES cells (Fig. 9A), indicating that normal genomic imprinting is disrupted in both Vivo and Vitro ES cell lines at later passages. Another imprinted gene, Snrpn, also exhibited biallelic expression at later passages (data not shown). To compare the pluripotency of Vivo and Vitro ES cells, we assessed mRNA expression, specific to the three germ layers, by real-time RT-PCR. No significant differences were found for Cdx2 (trophectoderm), T (mesoderm), Gata6 (primitive endoderm), and Nestin (neural progenitor) (Fig. 9B). This result indicates that Vitro ES cells have the same differentiation capabilities as Vivo ES cells in an in vitro environment.
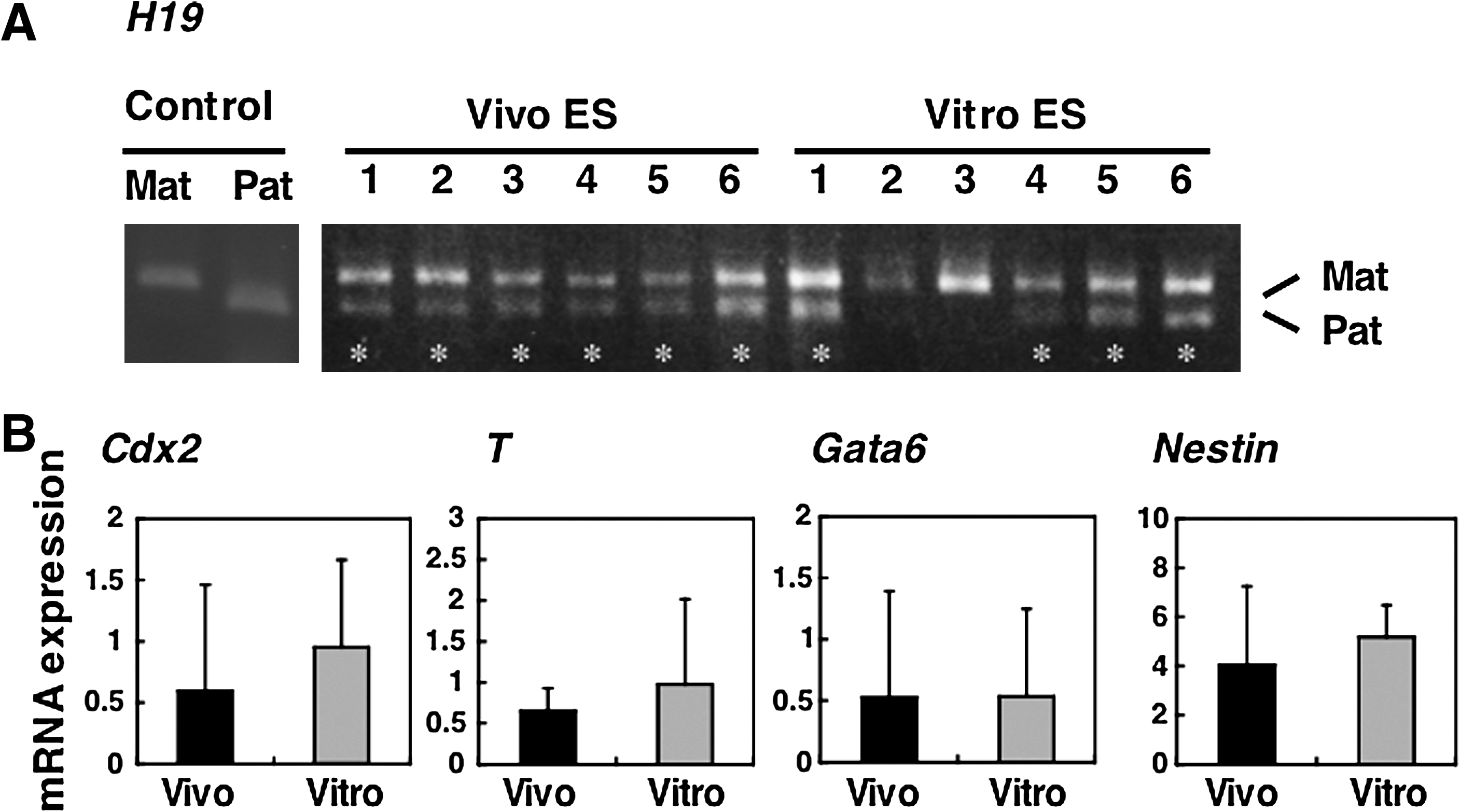
Gene expression patterns of Vivo and Vitro ES cell-derived EBs (BPF1 mouse strain). (
Discussion
Several studies have investigated the development of blastocysts in vitro; however, little is known about ES cell lines generated from these blastocysts. In humans, the collection of blastocysts in vivo is not an option due to ethical issues; thus, the only alternative is to obtain blastocysts through the techniques of IVF, IVC, and other reproduction technologies. In this study, we have compared the methylation status of imprinted genes and gene expression patterns in mouse ES cell lines derived from freshly collected in vivo blastocysts and from blastocysts cultured in vitro.
The expression of Oct3/4 in ES cells is essential for the maintenance of pluripotency. The loss of Oct3/4 expression has been correlated with a progressive loss of pluripotency upon spontaneous differentiation. The level of Oct3/4 mRNA is significantly higher in freshly collected in vivo blastocysts compared to blastocysts cultured in vitro (Tielens et al., 2006). Higher levels of Oct3/4 expression in blastocysts developed in vivo leads to a higher efficiency of ES cell line establishment when compared to the establishment of ES cell lines from blastocysts cultured in vitro (Tielens et al., 2006). However, in our study, we found no significant differences in establishment efficiency between Vivo and Vitro ES cells.
One of the reasons for similar establishment efficiencies may be that we used chemically defined knockout serum replacement (Knockout SR) for the establishment of ES cells in this study. In general, fetal bovine serum (FBS) is used as a component of the culture medium for ES cell establishment; however, FBS contains undefined factors, which occasionally stimulate the differentiation of ES cells (Horii et al., 2003). Therefore, the establishment of ES cell lines from blastocysts cultured in vitro is difficult if using FBS supplemented medium, because blastocysts with low levels of Oct3/4 mRNA expression are more easily differentiated by undefined factors (Tielens et al., 2006). In contrast, Knockout SR does not appear to contain these factors, and even if expression levels of Oct3/4 are low, ES cells can be generated from in vitro blastocysts as well as in vivo blastocysts, leading to a higher efficiency of establishment for Vitro ES cells than with FBS-supplemented medium. Despite the high establishment efficiency of the Vitro ES cells, the mRNA expression levels of the Oct3/4 gene were still lower in early passage (passage 2) Vitro ES cells than in Vivo ES cells, although the protein expression levels were the same as judged by immunoblot analysis. The difference between mRNA and protein expression data could be explained by the small difference in the mRNA expression data, which would have been difficult to detect by immunoblot analysis. In contrast, at later passages (passage 5 or 6), no significant differences in the expression levels of ES cell marker genes between in Vivo and Vitro ES cells were detected. We consider that ES cells with higher expression levels of ES cell marker genes might be able to survive more passages than ES cells with lower expression of these genes.
Next, we focused on genomic imprinting in ES cells. In general, imprinted genes are stably maintained during cellular division and differentiation (Pfeifer, 2000). In contrast, normal imprinting can be easily disrupted during preimplantation development in vitro. For example, in vitro cultured blastocysts often exhibit hypomethylation of the H19 DMR, which results in the biallelic expression of the H19 gene (Doherty et al., 2000; Mann et al., 2004; Sasaki et al., 1995). In our study, abnormal imprinting was still observed in early-passage Vitro ES cells. According to the expression analysis at this time point, Dnmt3b mRNA expression levels were significantly lower in Vitro ES cells than in Vivo ES cells. Dnmt3b is well known as a de novo methyltransferase, and is required for the establishment of new methylation patterns during embryonic development (Okano et al., 1999). Dnmt3b is also required for the maintenance of DNA methylation in addition to the major maintenance methyltransferase Dnmt1 (Dodge et al., 2005). These reports suggest that low expression levels of Dnmt3b may result in unstable genomic imprinting in Vitro ES cells.
In addition, hypomethylation in XX ES cells is associated with reduced levels of Dnmt3a and Dnmt3b, and it is known that the ectopic expression of these factors restores global methylation levels (Zvetkova et al., 2005
In any case, we observed several differences in methylation status and mRNA expression patterns between Vivo and Vitro ES cells at very early passages, but no significant differences were found at later passages. Long-term culture of ES cells often affects the methylation status of imprinted genes and their totipotency (Dean et al., 1998). In our study, obviously abnormal genomic imprinting appears in both Vivo and Vitro ES cell lines at passage 5. The H19 gene shows monoallelic expression in normal embryos (Mann et al., 2004); however, biallelic expression of this gene was observed at later passages of both types of ES cell line. We generated EBs to elucidate the pluripotency of Vitro ES cells, but we found no significant differences between Vivo and Vitro ES cells.
Our study has clarified that Vivo ES cells exhibited a more normal epigenotype than Vitro ES cells only at very early passages (below passage 2). In contrast, there was no significant difference between Vivo and Vitro ES cells at later passages. Therefore, we have to consider that repeated passages of ES cells disrupt normal genomic imprinting, leading to the abnormal biallelic expression of imprinted genes. Epigenetic alterations that arise after establishment and culture of ES cell lines are not corrected during postimplantation development, and these alterations are associated with aberrant imprinted gene expression in the fetus (Dean et al., 1998). In conclusion, it is advisable to use early-passage Vivo ES cells whenever possible to avoid abnormalities caused by long-term culture in mice. On the other hand, because Vitro ES cells are the only ones available in humans, a careful selection of ES cell lines is necessary to avoid the aberrant expression of imprinted genes, which could lead to abnormal development and disease.
Footnotes
Acknowledgments
We thank Dr. Tsukasa Oda (Gunma University) and Tomoyuki Tsukiyama (Kyoto University) for technical advice on immunoblot analysis, and Dr. Shoji Tajima (Osaka University) for providing the anti-Dnmt3b antibody. This work was supported in part by grants from the Japan Society for the promotion of Science (JSPS; 17770182); the Japan Science and Technology Corporation (JST); the Ministry of Education, Culture, Sports, Science and Technology of Japan; the Ministry of Health, Labour and Welfare of Japan; the Japan Health Sciences Foundation; and the National Institute of Biomedical Innovation.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.