
Abstract
Abstract
The generation of embryonic stem cell (ESC) lines from parthenogenetically activated oocytes can provide transplantable cells, which are immunocompatible for the oocyte donors as well as an invaluable tool for genetic engineering and epigenetic studies. We report the efficient isolation of eight putative bovine parthenogenetic embryonic stem cell (bpESC) lines from 15 in vitro produced parthenotes. Five of these cell lines were maintained for more than 15 passages (>140 days) and analyzed. The cells displayed typical ESC morphology, stained positive for alkaline phosphate by histochemical staining, expressed Oct4, Nanog, and either stage-specific embryonic antigens, SSEA1, or SSEA4, detected by immunofluorescence staining. RT-PCR analysis of the cells demonstrated expression of Oct4, Rex1, SSEA1, and ALP. All the cell lines except one had a normal karyotype of 60, XX. The cells differentiated in suspension culture to form embryoid bodies (EBs) expressing markers of the three embryonic germ layers as assessed by RT-PCR. In conclusion, we report efficient derivation of putative ESCs from bovine parthenogenetic embryos. The cells express pluripotent markers, have the ability to form EBs, and differentiate into cells of the three embryonic germ layers. This is the first report of characterized putative parthenogenetic bovine ESC lines.
Introduction
In livestock, this technology can be used in many ways: (1) to produce pharmaceutical proteins in blood or milk (Wilmut and Whitelaw, 1994); (2) to induce disease resistance traits against conditions such as mastitis in cows (Wall et al., 2005); (3) to knock out specific genes, for example, to make animals resistant to prion induced disease (Kuroiwa et al., 2004). In livestock, which have a long generation time compared with mice, we propose the ability to introduce transgene expression at specific sites or in specific tissues, may prove extremely valuable. In addition, such approaches would circumvent the scientific and ethical issues, which can arise from whole animal transgenesis. However, for this to be effective, transplantation of genetically modified cells will need to be achieved with robust contribution and colonization of the target tissues without fear of immune rejection by the recipients.
Immune rejection of transplanted cells by the recipient is mainly due to incompatibility of the major histocompatibility complex (MHC) antigens and immune acceptance depends on a graft that can be fully matched with respect to the phenotype of MHC subunits (Lin et al., 2003). Methods proposed to circumvent this problem include derivation of autologous ESCs from somatic cell nuclear transfer (SCNT) (Colman and Kind, 2000), or parthenotes (Cibelli et al., 2002; Vrana et al., 2003).
Parthenogenesis refers to embryonic development of eggs activated artificially without the presence of the male gamete. Parthenote cell lines could facilitate autologous tissue specific gene modification for recipient animals aimed at biofarming applications. In addition, they can provide valuable insights into developmental biology, including epigenetics.
The first parthenogenetic embryonic stem cells (pESCs) were isolated from mouse parthenotes 25 years ago (Kaufman et al., 1983), followed by a nonhuman primate (Cibelli et al., 2002), rabbits (Fang et al., 2006), and humans (Kim et al., 2007; Revazova et al., 2007). In the present study we derived and characterized putative bovine parthenogenetic embryonic stem cell (bpESC) lines. Given the commercial importance of the bovine in agriculture and as a large animal model for research, these cells provide important tools to improve animal and human health and to produce animals with a desirable genetic composition.
Materials and Methods
All reagents were purchased from Sigma Chemical Company (St. Louis, MO, USA) unless indicated in the text.
Experimental procedures were carried out under the guidelines of the Monash University Animal Ethics Committee and conducted according to the International guidelines for Biomedical Research Involving Animals.
Generation of parthenotes and in vitro fertilized (IVF) blastocysts
Ovaries were collected and transported from a local abattoir in prewarmed 39°C 0.9% (w/v) saline containing 0.05% (w/v) penicillin/streptomycin. They were washed with 0.9% (w/v) saline solution on arrival at the laboratory. Follicular fluid containing cumulus–oocyte complexes (COCs) were aspirated using an 18-G needle attached to a vacuum system. Follicular fluid was centrifuged at 600 × g for 1 min. The supernatant was discarded and the pellet containing COCs and other follicular cells was resuspended in HEPES-buffered Tissue Culture Medium (TCM-H; PH 7.4) which consisted of 10% (v/v) TCM 199 (10×), 2.0 mM NaHCO3, 2.0 mM pyruvic acid, 25.0 mM HEPES sodium salt, 1.0 mM L-Glutamine, 100 IU/100 μg/mL penicillin/streptomycin plus 30 IU/mL heparin (Pharmacia and Upjohn; 9041-08-1). Oocytes surrounded by two to three layers of cumulus cells were collected and washed in TCM-H. Groups of 30 COCs were transferred to individual wells of a four-well plate (Nalge Nunc International, Rochester, NY, USA) containing 600 μL in vitro maturation medium, which consisted of TCM-199 supplemented with 5 μg/mL luteinizing hormone (Lutropin-V, Bioniche Animal Health, Belleville, Ontario, Canada), 1 mM l-glutamine, 1 μg/mL β-estradiol, 100 IU/100 μg/mL penicillin–streptomycin and 10% (v/v) FBS. COCs were matured by culturing them at 39°C in a humidified gas environment of 5% CO2, 5% O2, and 90% N2 in air.
After 22 to 24 h of culture, cumulus cell were removed by vortexing (Thermoline Scientific, NSW, Australia) in handling medium, TCM-H plus 5% (v/v) FBS (JRH, Bioscience, Braybrook, Vic, Australia) containing 2 μg/mL hyaluronidase for 3.5 min. Metaphase II oocytes with a first polar body and normal morphology were chosen for parthenogenetic activation. Oocytes were activated in TCM-H containing 5μm calcium ionophore A-23187 for 4 min followed by three washes in TCM-H supplemented with 20% (v/v) FCS. They were subsequently transferred to preequilibrated synthetic oviductal fluid (SOF) medium (Tervit et al., 1972) with some modifications, including 107.5 mM-NaCl, 7.16 mM-KCl, 1.19 mM-KH2 PO4, 0.75 MgSO4, 6 μL/mL Lactic acid, 25 mM-NaHCO3, 0.03 mM-Phenol red, 0.727 mM pyruvic acid, 1.75 mM-CaCl2·2H2O, 8 mg/mL bovine serum albumin-BSA (γ-irradiated, fatty acid free-ABIVP; ICPbio, Auckland, New Zealand) supplemented with 75 × essential amino acids, 50 × nonessential amino acids, 100 μg/mL tri-sodium citrate dehydrate (Merck, Whitehouse Station, NJ, USA), 500 μg/mL myo-inositol, 2 mM L-glutamine, 50 μg/mL gentamycin plus 2 mM 6 dimethylaminopurine (6-DMAP) for an additional 3 h. The oocytes were washed three times in the modified SOF medium without 6-DMAP and cultured in modified SOF medium in four-well dishes with 400 μL mineral oil overlay in a humidified gas environment at 39°C of 5% CO2, 5% O2, and 90% N2 for 7–8 days. To produce IVF embryos following maturation, COCs were cocultured with bovine spermatozoa (2 × 10 6) in modified Fert-Talp medium at 39°C in a humidified gas environment of 5% CO2, 5% O2, and 90% N2 for 24 h (Daniels et al., 2001). Putative zygotes were then cultured in modified SOF medium at 39°C in a humidified gas environment of 5% CO2, 5% O2, and 90% N2 for 7–8 days. Development was assessed using a stereo microscope (Olympus I × 70) and embryos were photographed up to day 7.
Isolation and culture of putative bovine pESCs
The zona pellucidae of blastocysts were removed mechanically on day 7 and 8 using 27-G needles attached to 0.5 mL syringes (Insulin Needle, BD, Sparks, MD, USA). The zona free blastocysts were pressed onto mouse embryonic fibroblast (MEF) feeder layers, which were inactivated with mitomycin C. Pressed blastocysts were cultured at 39°C in a humidified gas environment of 5% CO2 in air in N2B27-3i medium containing N2B27 medium (an equal volume of DMEM/F12 + glutamax (Gibco, Gaithersburg, MD, USA) and Neurobasal medium (Gibco) with 1% (v/v) N2 and 2% (v/v) B27 supplements (Gibco) (Ying et al., 2003) plus three inhibitors (3i), 0.8 μM PD184352 (MEK 1/2 inhibitor, Stem Cell Sciences, Cambridge, UK), 2 μM SU5402 (FGF receptor inhibitor, Calbiochem, Gibbstown, NJ, USA) and 3 μM CHIR99021 (GSK3 inhibitor, Stem Cell Sciences) (Ying et al., 2008). This was designated as passage zero (P0). The medium was changed every other day. After 12 days of culture, the initial outgrowths from bovine parthenotes were mechanically dissociated into clumps and three to four pieces from each individual colony were replated onto fresh MEF feeder layers in Center-Well Organ Culture Dish (Becton Dickinson, Franklin lakes, NJ, USA) and designated as P1. Some clumps of 200–300 cells were also collected for RT-PCR. The growth of colonies was evaluated on a daily basis, with the cells passaged every 7 to 10 days. Each dish was specified for individual embryonal outgrowths and labeled Ex−Py, where E is the embryo and its specific number, Py to the passage number y. We have previously derived putative ESC lines from fertilized bovine embryos (Nadine Richings, personal communication). Some of these cell lines were used for comparisons with putative bpESC lines derived in this study.
Characterization of putative bpESC lines
Alkaline phosphatase staining
Alkaline phosphatase (ALP) activity was localized by histochemistry according to manufacturer's instructions using Alkaline Phosphatase Detection kit (Chemicon, Tumecula, CA, USA). Briefly, fast Red violet was mixed with Naphthol AS-BI phosphate solution and water in a 2:1:1 ratio. Cells were fixed for 15 min in 4% (w/v) paraformaldehyde at room temperature and then stained for a further 15 min. The cells were washed with PBS and analyzed for epifluorescence using an Olympus I × 71 microscope.
Immunocytochemistry staining
The putative bpESC lines were characterized by immunocytochemical methods using fluorescence-labeled antibodies against markers of undifferentiated bESCs following fixation of cells with 4% (w/v) paraformaldehyde at room temperature for 15 min. The cells for Oct4 and Nanog staining were permeabilized in 0.1% (v/v) Triton X-100 in 3%(v/v) goat serum in DPBS for 20 min. The putative bpESCs were incubated with 3% (v/v) goat serum in DPBS at room temperature for 1 h to block nonspecific binding of the primary antibodies and then incubated with primary antibodies raised against SSEA1 (Chemicon-Millipore, MAB4301), SSEA4 (Chemicon-Millipore, MAB4304), and Oct4 (Santa Cruz, Santa Cruz, CA, USA, sc-5279) and Nanog (Abcam, Cambridge, MD, USA, ab21603) diluted at 1:100 in DPBS containing 3% (v/v) goat serum overnight at 4°C. The next day, all colonies were washed with DPBS three times and incubated with secondary Ab (diluted in DPBS 1:1000, AlexaFlour 594 or 488, Invitrogen, Carlsbad, CA, USA) for 1 h at room temperature. After three washes with DPBS, cells stained with 1 μg/mL Hoechst 33342 (Sigma-Aldrich) in DPBS for 10 min at room temperature. Control cell lines were treated by omitting the primary antibodies (negative control). The cells were analyzed using an Olympus I × 71 microscope. Wavelengths used for excitation were 420–440, 470–490, and 520–550 for blue, green, and red, respectively, and for emission were 475, 510–550, and 580 for blue, green, and red, respectively.
RNA extraction and RT-PCR analysis of gene expression
Gene expression analyzed by RT–PCR is shown in the Table 1{TBL1}. Clumps of 200–300 cells were stored in 10 μL lysis buffer at −80°C until processing. Total RNA was extracted from samples using Dynabeads mRNA DIRECT Micro Kit (Invitrogen) according to the manufacture's instruction. Reverse transcription was performed using 20 μL RT system; 1 μL Superscript III Reverse Transcriptase (Invitrogen), 4 μL 5* First-Strand Buffer, 1 μL DTT, 1 μL dNTP Mixture (10 mM), 1 μL random primers (10 μM), 1 μL RNA inhibitor, 7.5 μL H2O, and 3.5 μL sample. A negative control without Superscript III was prepared to check for contamination by genomic DNA. PCR was performed in a Mycycler Thermal Cycler (Bio-rad, Hercules, CA, USA) at 50°C for 60 min, then 70°C for 15 min. The first-strand cDNA was further amplified by PCR using forward and reverse primers for specific genes. All samples were checked for β-Actin (housekeeping gene) to verify the success of the RT reaction and then for other specific genes with individual primers. PCR amplification was performed in 25 μL reaction containing 2.5 μL DNA polymerase 10× reaction buffer, 1.5 μL MgCl2 (25 mM), 0.5 μL dNTP Mixture (10 mM), 0.2 μL Taq DNA Polymerase, 1 μL (10 μM) from each forward and reverse primers, 1 μL sample, and 17.3 μL H2O (All chemicals were purchased from the Fisher Biotec, Wembley, Western Australia, Australia).The prepared solution was processed in a Mycycler Thermal Cycler and run for 35 cycles; denaturation (95°C, 45 sec), annealing (55–56°C) and extension (72°C, 45 sec) steps.
All PCR samples were analyzed by electrophoresis on a 2% (w/v) agarose gel (Bioline) containing 4% (w/v) ethidium bromide. The sequence of primers used for PCR and the product size are listed in the Table 1.
Karyotyping of putative bpESC lines
Karyotyping was performed at P6–P11. To estimate chromosome number the cultures were treated with 5-bromo-2-deoxyuridine (BrdU) overnight and then with Colcimid (Gibco) for a further 4 h to suppress mitosis. After treating with TrypLE Express (Invitrogen) and hydrating in hypotonic KCL for 15 min, they were washed and fixed in methanol and acetic acid (BDH, Bristol, UK; 100015N) in a ratio of 3:1. The fixed cells were dropped on to clean slides at RT. The slides were stained with a freshly made staining solution containing 3 mL of Leishman stain in 17 mL Gurrs buffer (Invirogen) for 8 min. The Leishman stain was prepared by dissolving 2 g Leishman powder in 1 liter methanol. A coverslip was mounted on the slides with Histomount (National Diagnostics, Atlanta, GA, USA; HS-103) and slides viewed using a light microscope under oil immersion optics (Nikon C1) at 1000 × magnification.
Differentiation potential of putative bpESC lines
The putative bpESCs colonies were mechanically dissociated into clumps of 200–300 cells with 27-G needles and cultured in low adhesion culture dishes (Corning Incorporated, Corning, NY, USA) in medium containing α-Minimum Essential Medium (α-MEM, with deoxyribonuceosides and ribonucleoside; Gibco, Invitrogen Australia, Mount Waverley, Victoria, Australia), 2 mmol/mL Glutamax (Gibco), 0.1% (v/v) Mercaptoethanol (Gibco), 1% (v/v) nonessential amino acid (NEAA, Gibco), 1% (v/v) ITS (10 μg/mL insulin, 5.5 μg/mL 125 transferrin, 6.7 ng/mL selenium; Gibco), 0.5% (v/v) penicillin–streptomycin (Gibco), 20% (v/v) FBS at 39°C in a humidified gas environment of 5% CO2 in air. Culture medium was changed every 2 to 3 days. Samples from attached and nonattached EBs were collected at 30 and 72 days to check gene expression of ectodermal markers (Pax6, Vimentin, β-3-Tubulin, Nestin), endodermal markers (Gata6, Somatostatin, Transthyretin-TTR), and mesodermal markers (Connexin40, BMP4) (Table 1) by RT-PCR as described before.
Results
Generation of parthenotes
In a replicated experiment a total of 113 COCs were incubated at 39°C in a humidified gas environment of 5% CO2, 5% O2, and 90% N2 in air. The maturation rate of oocytes to metaphase II stage was 42.5% (48 of 113). Seventy-three percent (35 of 48) of activated embryos cleaved within 38 h (Table 2). From days 2–5, the embryos divided without a change in overall size of the embryo, as the cells of the embryo became smaller. Morphology of parthenotes was comparable to IVP embryos during all stages of development (Fig. 1). On approximately day 5 at the morula stage, the junctions between the cells becomes tight and embryos started compacting (Fig. 1D). Blastulation or cavitation commenced around day 6 with fluid accumulating in the blastocoelic cavity with the trophectoderm and inner cell mass being readily apparent (Fig. 1E). The inner cell mass of parthenotes was similar morphologically and in size to that of IVF embryos at the same stage.

Development of a typical parthenote embryo (
Three 3 embryos used for preliminary experiments.
One line was lost due to technical problems.
Isolation and culture of putative bpESC lines
Primary outgrowths forming dome and spherical shapes with well-defined boundaries, grew over the top of the MEF feeder layer within 12 days. Eight putative bpESC lines were derived. About 50% of replated pieces from each embryo at P2 and P3 did not give outgrowths. E3 did not grow at P3 and E6 at P2. However, after passage 3 more than 90% of replated pieces expanded and formed dome and spherical colonies and in the later passages (after P5) putative bpESC lines were similar in morphology to ESC lines derived from bovine IVF embryos (Fig. 2). Six putative bpESC lines were established and five lines were maintained for more than 15 passages (>140 days) (Table. 2).
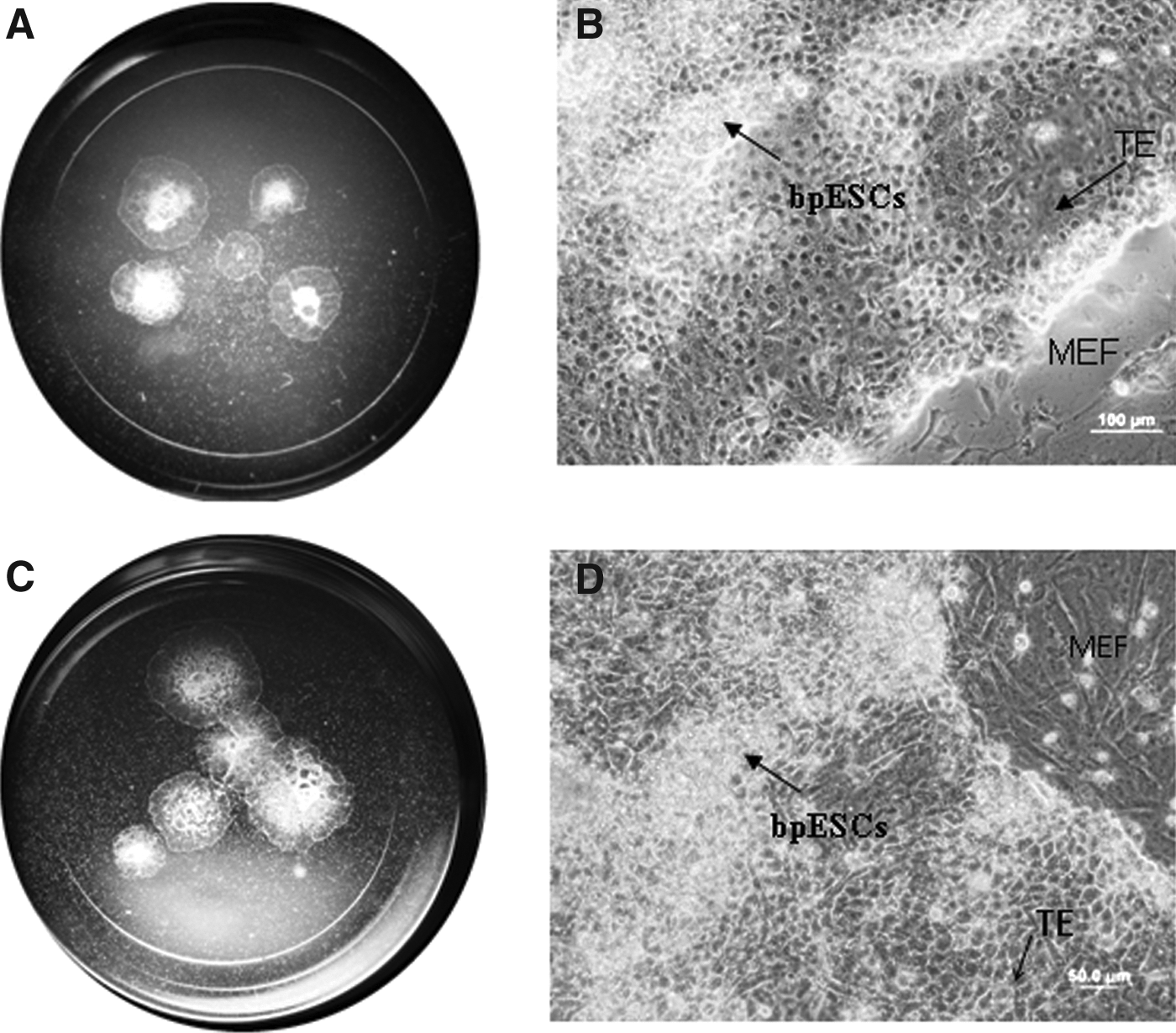
View of an organ culture well containing explants of (
Characterization of putative bpESC lines
Putative bpESCs appeared very morphologically similar to putative bovine ES cells isolated from IVF embryos. The center of colonies contained more ES cells with large numbers of lipid inclusions (Fig. 2A) and the edge of the outgrowth resembled trophectoderm-like cells, which were of a large size but with few lipid inclusions. When the colonies expanded, the cells with a large numbers of lipid inclusions were compacted in different sections of the colony and were seen as white shiny spots under the dissecting microscope (Fig. 2B).
The five surviving lines were characterized by molecular analysis. MEFs were negative for Oct4, Rex1, SSEA1, and ALP by RT-PCR, while the putative bpESC were positive (Fig. 3A). RT-PCR analysis showed mRNA expression of key pluripotent markers including Oct4, Rex1, SSEA1, and ALP not only at early passages, P0–P1 (Fig. 3B) but also all five cell lines showed expression of Oct4, Rex1, and SSEA1 in later passages, P11 (Fig. 3C). All lines expressed a high level of alkaline phosphatase activity, at P7–P8 (Fig. 4A–C) and at P15. Each of the cell lines expressed SSEA1 or SSEA4 at P4–P8, as determined by immunofluorescent staining (Fig. 4D–I). The cells were positive for Oct4 protein (Fig. 4J–L) and Nanog protein (Fig. 4M–O) at P15 as demonstrated by immunofluorescent staining. Bovine fibroblasts were confirmed to be negative for ALP staining and protein expression of Oct4, Nanog, SSEA1, and SSEA4 by immunohistochemistry (data not shown).
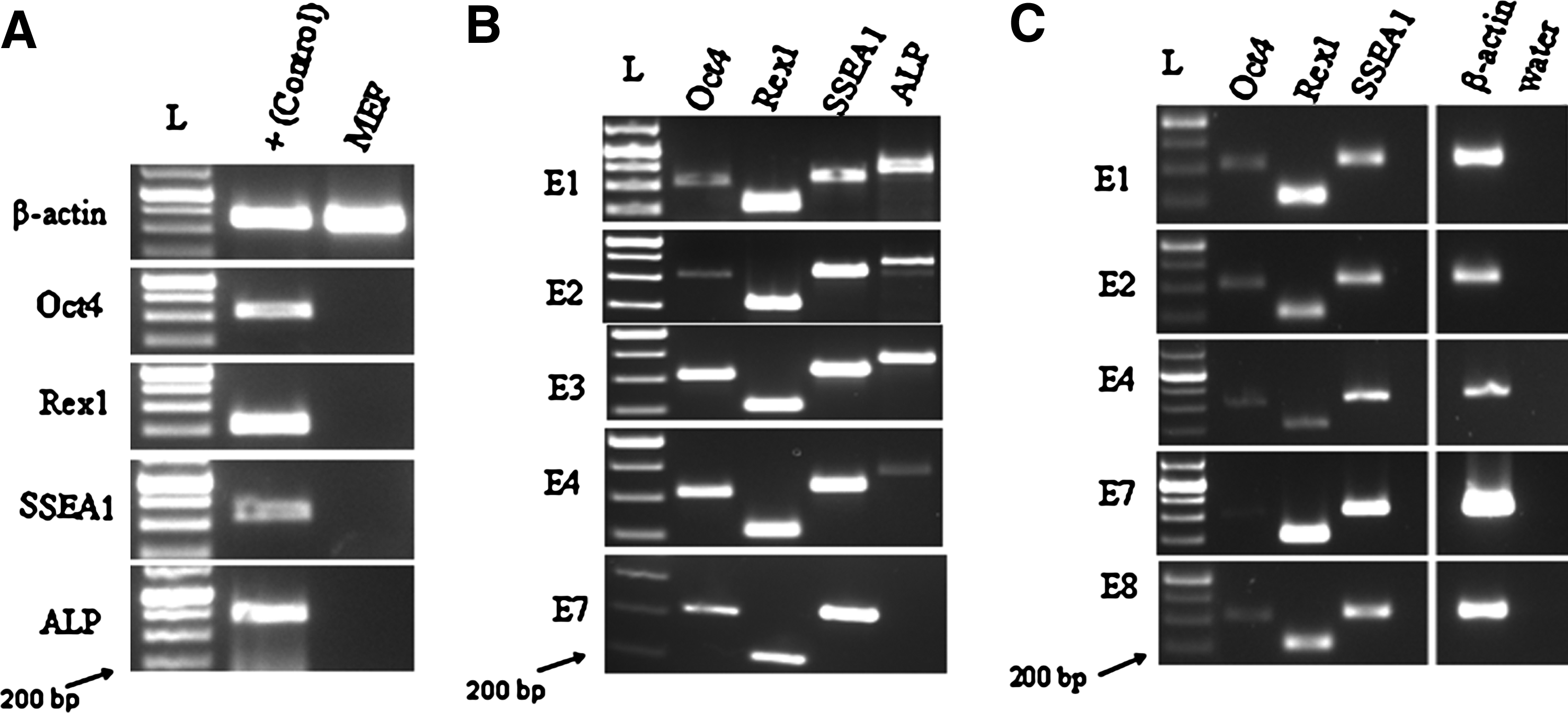
Five cell lines were checked for β-actin expression (data not shown) and then analyzed for pluripotent markers. (
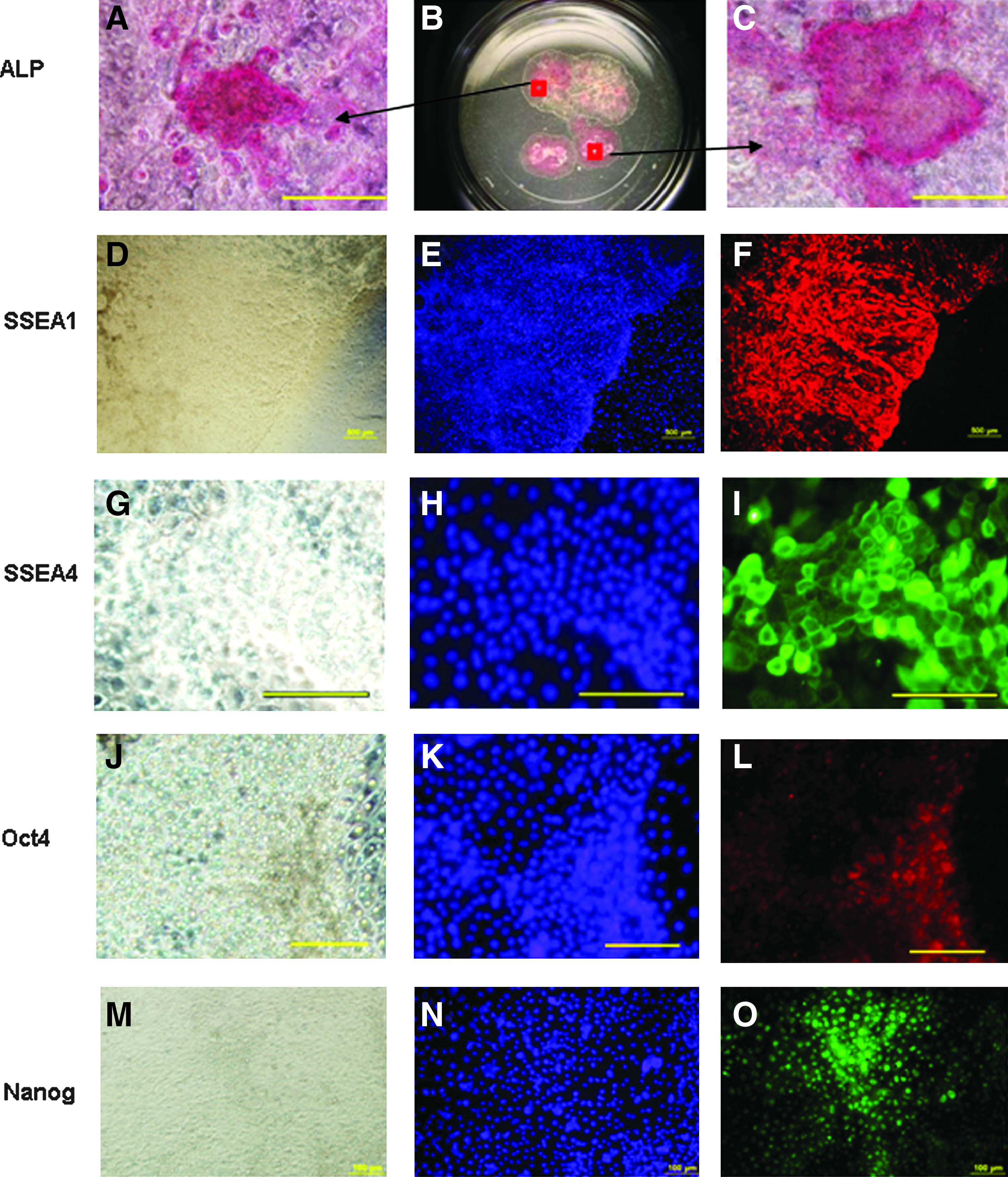
Alkaline phosphatase-positive staining of putative bpESC lines colonies with (
Karyotyping of putative bpESC lines
Four of five cell lines examined had normal karyotype with 60–90% of cells in spreads having a chromosome count of 60, XX (Fig. 5A). An abnormal chromosome count of 62, XX was observed in the majority of cells in line E1 at P10.

Normal karyotype, embryoid body (EB) formation and analysis of gene expression. (

Differentiation potential of putative bpESC lines
Cystic and noncystic EBs (Fig. 5B) formed from putative bpESCs over 5 days in suspension culture and after 15 days they attached onto a dish and developed outgrowths. RT-PCR results demonstrated mRNA expression of genes representative of the three embryonic germ layers ectodermal markers: Pax-6, β-3-Tubulin, Nestin, Vimentin; endodermal markers: Somatostatin, Transthyretin (TTR), and Gata6; and mesodermal markers: Connexin40 and BMP4. The expression of pluripotent markers, Oct4, Rex1, and ALP except a weak band of SSEA1 was not detected in EBs (Fig. 5C).
Discussion
This article describes the generation of bovine parthenogenetic embryos, the assessment of in vitro development of parthenotes, and the efficient isolation, derivation, and preliminary characterization of putative pbESC lines. Diploid bovine parthenotes were produced by activation of metaphase II oocytes by exposure to calcium ionophore and prevention of extrusion of the second polar body by subsequent 6-DMAP treatments. Development of parthenotes to the blastocysts stage was similar to IVF embryos. The inner cell mass of parthenotes was similar morphologically and in size to that of IVF embryos at the same stage; however, the trophectoderm cells in parthenote embryos were larger than in IVF embryos.
Several groups have reported derivation of bovine ES-like cell lines from in vitro produced blastocyst-stage embryos (Cao et al., 2009; Cibelli et al., 1998; Gjorret and Maddox-Hyttel, 2005; Iwasaki et al., 2000; Kitiyanant and Saikhun, 2000; Mitalipova et al., 2001; Pant and Keefer, 2009; Saito et al., 1992, 2003; Wang et al., 2005; Yadav et al., 2005), and there is one preliminary report on putative bovine parthenogenetic ESC isolation by Roach et al. (2006); however, there is no report of robust generation of well characterized bpESC.
We established six putative ES cell lines from bovine parthenotes. Putative bpESCs proliferated in a manner similar to IVF ESC lines but tended to differentiate more frequently at early passages. However, once established, parthenote cell lines maintained ESC characteristics. The one cell line that was lost was due to technical problems not related to differentiation or cell death.
Characterization of the five lines demonstrated their pluripotentiality after long-term culture. Putative bpESCs shared certain characteristics with putative IVP-ESCs, for example, high alkaline phosphatase activity, the expression of pluripotent markers including SSEA1, SSEA4, Oct4, and Rex1. Further, the cells demonstrated EB formation and differentiation potential, however, isolation of putative bpESC lines was less efficient than putative IVF ESC lines with fewer bpESCs expressed Oct4 protein than putative IVF ESCs (Nadine Richings, personal communication).
The detection of Oct4 alone may not be a definitive indicator of pluripotency as both ICM and TE express Oct4 in bovine blastocysts. The absence of Oct4 transcripts in the TE of the bovine day 7 blastocysts, despite Oct4 protein being detected suggests the transcripts may have been generated at or before the blastocyst stage and persisted for a few days (Kurosaka et al., 2004). In equine blastocysts Oct4 expression was observed in in vitro cultured blastocysts, whereas conversely, it was suppressed in embryos exposed to an in vivo environment (Choi et al, 2009). Moreover, Oct4 expression reduces after day 12 in bovine embryos and is localized to the pluripotent component (van Eijk et al., 1999; Gjorret and Maddox-Hyttel, 2005). Despite the conflicting reports on the expression of Oct4 in the trophectoderm of livestock species, it is expressed in the ICM and hence expected to be expressed in ESCs. Therefore, we believe the prolonged expression of Oct4 is indicative of pluripotency as the pluripotent ES lines in this study have been maintained for ∼4 months in culture.
Rex1 may also play a role in the maintenance of pluripotency in bovine embryonic cells as it is expressed simultaneously with Oct4 in in vitro-produced bovine blastocyst (Vigneault et al., 2009). The use of other pluripotent markers including Rex1 would be very useful for such studies; unfortunately, the availability of bovine validated antibodies are limited.
Bovine pESCs can provide an important model system to study the effect of paternally imprinted genes on cell differentiation. Differentiation ability of mouse pESC lines that was analyzed by teratoma formation did not contribute to mesodermal linage particularly skeletal muscle (Allen et al., 1994). Therefore, the absence of paternally imprinted genes may contribute to limited differentiation potential.
We have shown that putative bpESC can also retain the capacity to express marker genes indicative of the three embryonic germ layers in vitro. This is similar to the data of Dighe et al. (2008) in the rhesus monkey and Revazova et al. (2007) in human parthenogenetic ESC lines.
Both parthenogenesis and nuclear transfer (NT) are potential strategies for producing histocompatible ES cells for therapeutic applications. Both nuclear and mitochondrial genomes of ESCs derived from parthenogenesis are the same as the oocyte donor, whereas NT-ESCs have only a nuclear match but not a mitochondrial match with the donor. Also, parthenogenesis is more efficient at producing embryos than SCNT (Kim et al., 2007).
Our findings indicate that histocompatible pluripotent ESC lines can be obtained with a high efficiency in large animal models. These autologous cells in addition to genetic engineering or therapeutic applications could provide a useful tool for investigation of epigenetic modifications.
Footnotes
Acknowledgments
We thank Nadine M. Richings for her advice, David Galloway for critical comments on the manuscript, Kate Zang for designing primers, Paul Bello (Stem Cell Sciences) for providing N2B27-3i medium.
Author Disclosure Statement
The authors declare that they have no conflicting financial interests.