
Abstract
Abstract
Nuclear transfer is a very effective method for propagation of valuable, extinct, and endangered animals. Hand-made cloning (HMC) is an efficient alternative to the conventional micromanipulator-based technique in some domestic species. The present study was carried out for the selection of suitable somatic cells as a nuclear donor and development of an optimum culture system for in vitro culture of zona-free goat cloned embryos. Cleavage and blastocyst rates were observed 72.06 ± 2.94% and 0% for fresh cumulus cells, 81.95 ± 3.40% and 12.74 ± 2.12% for cultured cumulus cells, and 92.94 ± 0.91% and 23.78 ± 3.33% for fetal fibroblast cells, respectively. There was a significant (p < 0.05) increase in blastocyst production in goats when cultured on a flat surface (FS) (23.78 ± 3.33 %) than well of wells (WOW) (15.84 ± 2.12 %) and microdrops (MD) (0.7 ± 0.7%). Furthermore, cleavage and blastocyst production rates were significantly (p < 0.05) more in the WOW (15.84 ± 2.12%) than the MD (0.7 ± 0.7%) system. The quality of HMC blastocysts was studied by differential staining. Genetic similarity was confirmed by polymerase chain reaction (PCR)-based amplification of the second exon of the MHC class II DRB gene, which gave similar bands in electrophoresis (286 bp) both in cloned embryos and donor cells. In conclusion, the present study describes that the fetal fibroblast cell is a suitable candidate as nuclear donor, and the flat surface culture system is suitable for zona-free blastocyst development by the hand-made cloning technique in the goat.
Introduction
Goat is an important source of milk, meat, hair, and manure throughout the world and it helps in sustaining the economy of the farmers. In the past, several attempts have been made to produce cloned (Lan et al., 2006; Shen et al., 2006) and transgenic goats (Baguisi et al., 1999; Keefer et al., 2001) using the micromanipulator system. However, no information is available to date, on the application of a HMC technique in goats, except a study by Akshey et al. (2008).
Despite the widespread success of somatic cell cloning, this technology is being hampered by various biological and technical problems (Vajta et al., 2003). Dinnyes et al. (2002) suggested the need to evaluate the technical procedures and the biological materials to further improve the efficiency of cloned embryo production. Various factors, viz., treatment of the donor cells and recipient oocytes, are known to be critical for the successful outcome of this technology.
Since the landmark achievement of Wilmut et al. (1997), there has been constant debate among the researchers about the appropriate donor cell cycle stage and cell type to be used as a nucleus donor. Investigators have evaluated various types of donor cells, such as embryonic stem cells, blastomeres, fibroblast cells, etc., with varied success rate in producing the blastocysts (Keto et al., 1998). Therefore, suitable donor cell types need to be identified for improving the efficiency of producing developmentally competent blastocysts.
Additionally, the culture system is also an important factor that can affect the efficiency of blastocyst production using the HMC method. Several culture systems like well-of-the-wells (WOW) (Vajta et al., 2001), agarose gels (Peura and Vajta, 2003), glass oviduct (Thouas et al., 2003), and microdrops (Oback et al., 2003) have been developed in the past for zona-free cloned embryos with variance in efficiency of blastocyst production. However, not mamy attempts have been made to evaluate the suitability of donor cell types and culture conditions for zona-free reconstituted goat embryos. Therefore, the present study was undertaken to select the suitable donor cells and to develop an optimized culture system for the preimplantation development of zona-free cloned goat embryos.
Materials and Methods
All chemicals and media were purchased from Sigma Chemical Co. (St. Louis, MO, USA) and disposable plastic wares were procured from Falcon-ware Becton-Dickinson (Bedford, MA, USA) unless otherwise stated.
In vitro maturation of oocytes
In vitro maturation (IVM) of goat oocytes were carried out as described by Malakar and Majumdar (2005). Briefly, goat ovaries were collected from a local abattoir and transported to laboratory within 3 h of collection in a thermo flask containing 0.9% normal warm (32–35°C) sterile saline fortified with antibiotics (50 μgmL−1 gentamicin sulphate). The ovaries were washed four to five times with normal saline supplemented with antibiotics. Cumulus oocyte complexes (COCs) were aspirated by a puncturing method in the aspiration medium consisting of TCM-199 (HEPES modified) and bovine serum albumin (BSA) (0.3% w/v). COCs with more than three cumulus layers and homogeneous ooplasm were taken for in vitro maturation. COCs were washed two times with washing medium containing TCM 199 (HEPES modified), 10% fetal calf serum (FCS), 0.05 mgmL−1 sodium pyruvate, 0.0035 mg/mL−1 L-glutamine, and 50 μg/mL−1 gentamicin sulphate and three times with maturation medium containing TCM-199 (HEPES modified), 10 μg/mL−1 LH, 5 μg/mL−1 FSH, 1 μg/mL−1 estradiol-17β, 0.05 mg/mL−1 sodium pyruvate, 5.5 mg/mL−1 glucose, 35 μg/mL−1 L-glutamine, 50 μg/mL−1 gentamicin, 3 mg/mL−1 BSA, and 10% EGS (heat-inactivated estrous goat serum). COCs (15–20 oocytes) were placed in 100 μL of maturation medium, covered with mineral oil and incubated in a CO2 incubator (5% CO2 in air) at 38.5°C for 22 h with maximum humidity.
Preparation of recipient cytoplast
The recipient cytoplasts were prepared as described previously (Vajta et al., 2006) with minor modifications. COCs with expanded cumulus cells were removed from the maturation droplet 24 h postmaturation and pipetted vigorously in TCM-199 medium supplemented with 25 mM HEPES and 5% FCS to remove the cumulus cells. The denuded oocytes with evenly granular ooplasm were incubated with pronase (2 mg/mL−1) in T10 medium (where T denotes HEPES modified M-199 supplemented with 2.0 mM L-Glutamine, 0.2 mM Sodium Pyruvate, 50 μg/mL−1 gentamicin, and the following number denotes 10% FBS) for 8 min at 38.5°C. When the oocytes with completely digested zona pellucida were transferred to T20 medium (T medium containing 20% FBS) and incubated at 38.5°C until a prominent protrusion cone was observed. The oocytes showing a protrusion cone were transferred to a 35-mm dish containing T20 medium with 2.5 μg/mL−1 cytochalasin-B and mechanically bisected under a stereo zoom microscope (Olympus, Japan) using an ultrasharp splitting blade (SurgeEdgeTM) in such a way that the protrusion cone remains in the smaller half of the oocyte. The larger demioocytes with no protrusion cones were incubated in T20 medium for 15 min to regain the spherical shape.
Preparation of donor cells
Three types of somatic cells, viz., fetal fibroblast cells, fresh cumulus cells, and cultured cumulus cells, were evaluated to be used as the nucleus donor.
The source for fetal fibroblast cells were skin explants from goat fetuses of about 60 days collected from a local abattoir. The skin explants were washed seven to eight times with sterile Dulbecco's phosphate buffer saline (DPBS). Tissue slices were cultured using separate four-well dishes (4WD) (Nunc, Denmark) in Dulbecco's modified eagles medium (DMEM) supplemented with 20% FBS and 50 μg/mL−1 gentamicin sulphate in 5% CO2 at 37°C with maximum humidity. Monolayers of fibroblast cells were grown until confluency and were subcultured by partial trypsinization.
Cumulus cells were collected from in vitro matured COCs. After maturation, the expanded COCs were removed from the maturation droplet and pipetted vigorously in TCM-199 medium containing 25 mM HEPES and 5% FCS. The collected cumulus cells were cultured in 4WDs containing DMEM supplemented with 20% FBS at 37°C, 5% CO2 in air, with maximum humidity. After reaching confluence, the monolayer was trypsinized and the cells were passaged further.
The confluent cells (both fetal fibroblast and cumulus cells) at the fifth to sixth passage were allowed to grow, for another 3 days, in order to achieve overconfluence. As a result, the majority of the cells were expected to reach the G0/G1 stage of the cell cycle. The cells were trypsinized and collected in T20 medium in order to be used as a nucleus donor.
Pairing and fusion
The enucleated demioocytes were incubated in phytohemagglutinin (0.5 mg/mL−1) for 3–4 sec and transferred to T20 medium containing a low density of donor cells. These cells were prepared during the incubation period of 10–15 min to allow rounding up of the demioocytes. Each demioocyte was allowed to get attached to a single, rounded, medium-sized donor cell by phytohemagglutinin treatment for couplet formation. The couplets (demioocyte and donor cell pairs) were transferred to fusion medium containing 0.3 M Mannitol, 0.1 mM MgSO4, 0.05 mM CaCl2, and 3 mg/mL−1 BSA for equilibration. The couplets and the other enucleated demioocytes were transferred to northern and southern parts of the fusion chamber (BTX micro slide 0.5-mm gap, model 450; BTX, San Diego, CA, USA) containing the fusion medium. Electrofusion of the triplets (couplet–demioocyte sandwich) was then carried out by a single-step method. A demioocyte and a couplet were picked up using a fine pulled capillary pipette (Unopette® Becton Dickinson, Franklin Lake, NJ, USA) having an inner diameter of 100–120 μm. Initially, the couplet was expelled and aligned with an AC pulse (7 volts) using a BTX Electrocell Manipulator 2000 (BTX), so that the somatic cell faces the negative electrode. Immediately after alignment, another demioocyte was introduced into the fusion chamber close to the couplet. As soon as the somatic cell was sandwiched between the demioocytes, a double DC pulse of 2.31 kV/cm for 15 μsec was applied. The triplets were then incubated in T20 medium for rounding up and subsequent reprogramming for 4 h at 38.5°C in a CO2 incubator.
Activation and in vitro culture of the cloned embryo
The reconstructed oocytes were activated using 2 μM Ca ionophore in T20 medium for 5 min at 38.5°C after 4 h of fusion. The triplets were washed three times in T20 media and kept in individual droplets of 5 μL of T20 containing 2 mM 6-Dimethylaminopurine (6-DMAP) followed by incubation for 3 h in 5% CO2 in air at 38.5°C.
The activated embryos were washed with embryo development medium (EDM) containing TCM-199 (HEPES modified), 10% FCS, 10 μL/mL−1 essential and 5 μL/mL−1 nonessential amino acids (from Sigma Chemicals) and 10 mg/mL−1 BSA and cultured in three different culture systems: (1) WOW: hand-made microwells of 300 μm width and deepth were prepared with the help of a smooth, “V”-shaped darning needle (Booth et al., 2001) in a well of a 4WD (Nunc; Roskilde, Denmark) containing 400 μL of embryo development medium. This culture system is known as the WOW system (Vajta et al., 2001). The WOWs were prepared a day before the in vitro culture (IVC) of embryos, rinsed, and replaced with fresh medium just before IVC and covered with 400 μL of mineral oil. (2) Micro drops (MD): 5 μL droplets of EDM were prepared in a 4WD (10 microdrops in each well) and covered with 400 μL of mineral oil, (3) flat surface (FS): 400 μL of EDM was added to each well of the 4WD and covered with 400 μL of mineral oil and 10–15 presume cloned embryos were cultured in each well of 4WDs (FS). The embryos were kept carefully well apart in the periphery of the well of the 4WD to avoid aggregation or separation of single blastomere. The attachment of embryos was prevented due to high viscosity of culture medium owing to 10% FCS and 10 mg/mL−1 BSA.
Quality assessment of HMC embryos and determination of blastocyst cell number
Rates of embryo development in terms of cleavage and blastocyst formation were observed on day 7 of IVC, and percentage of development of each stage of embryo was determined. The health of the blastocysts was determined by counting the cell numbers by differential stainings as described by Thouas et al. (2001). The blastocysts were incubated in DPBS with 1% Triton X-100 and 100 μg/mL−1 propidine iodine for 15 sec and immediately transferred to 500 μL of fixative solution containing 100% ethanol with 25 μg/mL−1 Hoechst 33342 for overnight incubation at 4°C. The cell number was counted using an inverted microscope (Nikon) fitted with an UV lamp and excitation filters.
Confirmation for production of cloned embryos from donor cells
Genomic DNA was isolated from the cloned embryos and nuclear donor cells. The second exon of the goat major histocompatibility complex (MHC) class II DRB gene (286 base pair) was amplified using the primer pair (F: 5′TATCCCGTCTCTGCAGCACATTTC3 ′; R: 5′ATCGCCGCTGCACACTGAAACTC3 ′) suggested by Keefer et al. (2002). The PCR product from the cloned embryos and nuclear donor cells was further analyzed through SSCP to confirm their genetic identity.
Statistical analysis
The data was analyzed using SYSTAT 7.0 (SPSS Inc., Chicago, IL, USA) after arcin transformation. Differences between means were analyzed by one-way analysis of variance (ANOVA) followed by a Fisher's LSD test. Significance was determined at p < 0.05.
Results
Oocyte bisection method and enucleation efficiency
A prominent protrusion cone was observed on the surface of each oocyte (Fig. 1A) upon transfer and subsequent incubation of zona pellucida-free oocytes in T20 medium (Fig. 1A). The higher enucleation efficiency of >95% (380/400, 6 replicates), achieved in the present work through protrusion cone guided bisection and selection of enucleated demioocyte without Hoechst 33342 staining indicated this approach to be a superior one for enucleating the zona-free goat oocytes (Fig. 1B).
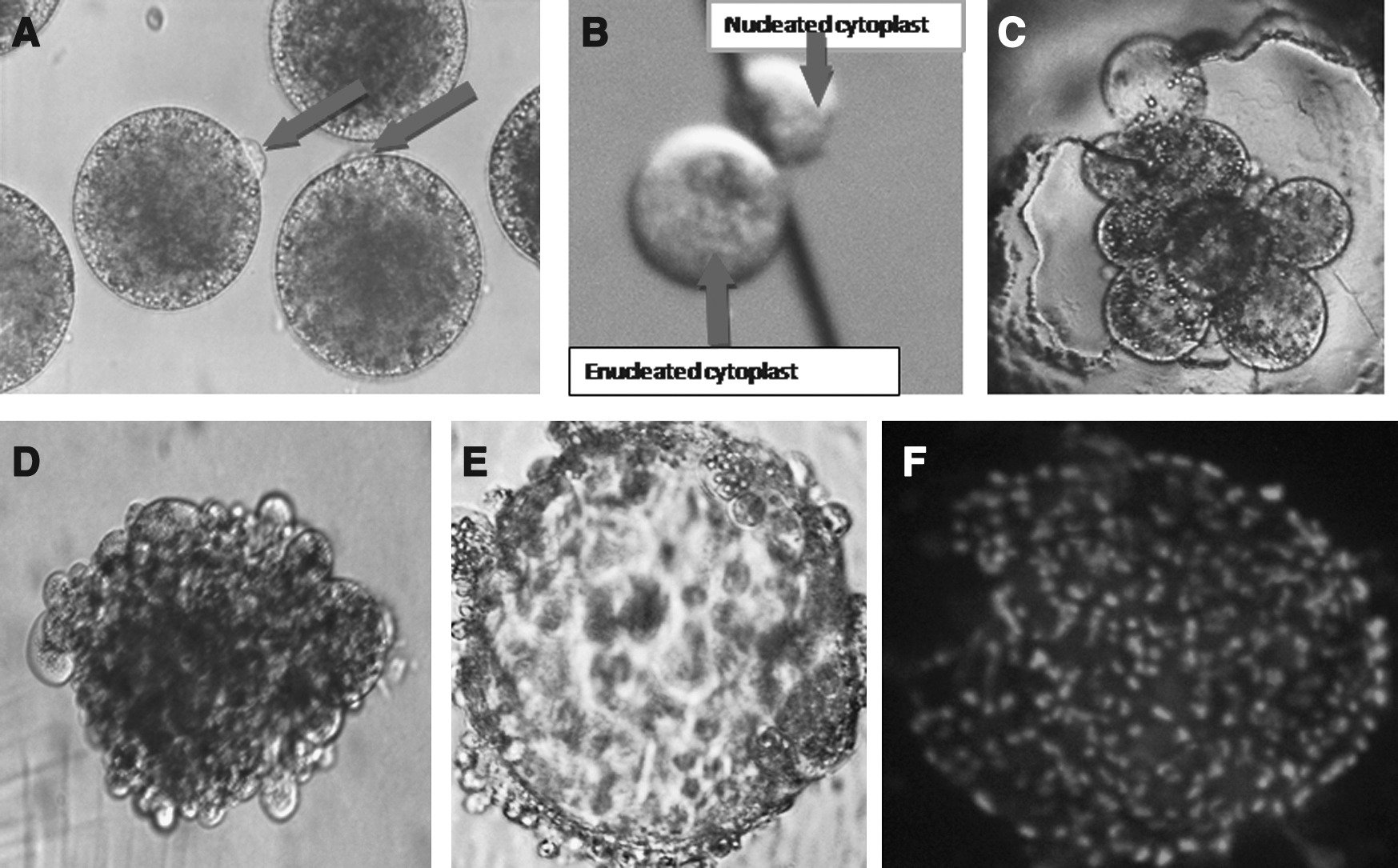
Protrusion cone (arrow) produced on the surface of zona-free IVM goat oocytes within 30 min in T20 medium. (
Experiment 1: Effect of different donor cells on development of hand-made cloned embryos
The three types of donor cells evaluated in the present study showed no significant difference in the couplet formation, triplet formation, fusion, cleaved embryo (Fig. 1C), and blastocyst (Fig. 1E) production rate. However, in terms of triplet formation and fusion rate, cultured cumulus cells provided relatively better result than fresh cumulus cells and fetal fibroblast cells (Table 1).
Figures quoted as percent mean ± SEM (n). Data from six trials for each donor cell type.
Values having different superscripts in the same column differ significantly (p < 0.05).
A significant (p < 0.05) increase in cleavage rate (92.94 ± 0.91%) and blastocyst production rate (23.78 ± 3.33%) was observed with fetal fibroblast cells compared to fresh cumulus cells (72.06 ± 2.94% and 0.00 ± 0.00%) and cultured cumulus cells (81.95 ± 3.40% and 12.74 ± 2.12%) (Table 1). Interestingly, no blastocyst production was seen with fresh cumulus cells, which might be attributed to the factors like composition of medium and/or biological materials.
Experiment 2: Effect of different culture systems on development of hand-made cloned embryos
Embryo development medium was used for comparing the development of cloned embryos in different culture systems. Considering the high cleavage and production rate observed, fetal fibroblast cells were used as nuclear donors for the present work. When compared, development and cleavage rates were better using a flat culture system (Table 2). Even in the case of zona-free reconstructed embryos, flat systems showed a significant (p < 0.05) increase in blastocyst production rate than the WOW or MD system (Table 2). Furthermore, cleavage and blastocyst production rate were significantly (p < 0.05) more in the WOW (15.84 ± 2.12 %) than the MD (0.7 ± 0.7%) system (Table 2).
Figures quoted as percent mean ± SEM (n). Data from six trials.Values having different superscripts in the same column differ significantly (p < 0.05).
FS, flat surface; WOW, well of the wells.
The Hoechst 33342 staining reflected the good quality of HMC blastocyst (∼155 cells/blastocyst; Fig. 1F). HMC embryos produced from donor somatic cells were confirmed by PCR amplification of the highly polymorphic second exon of MHC class II DRB (Fig. 2). The PCR-SSCP analysis of the 286-bp product revealed a similar pattern for both cloned goat embryos and donor cells, confirming their genetic identity.
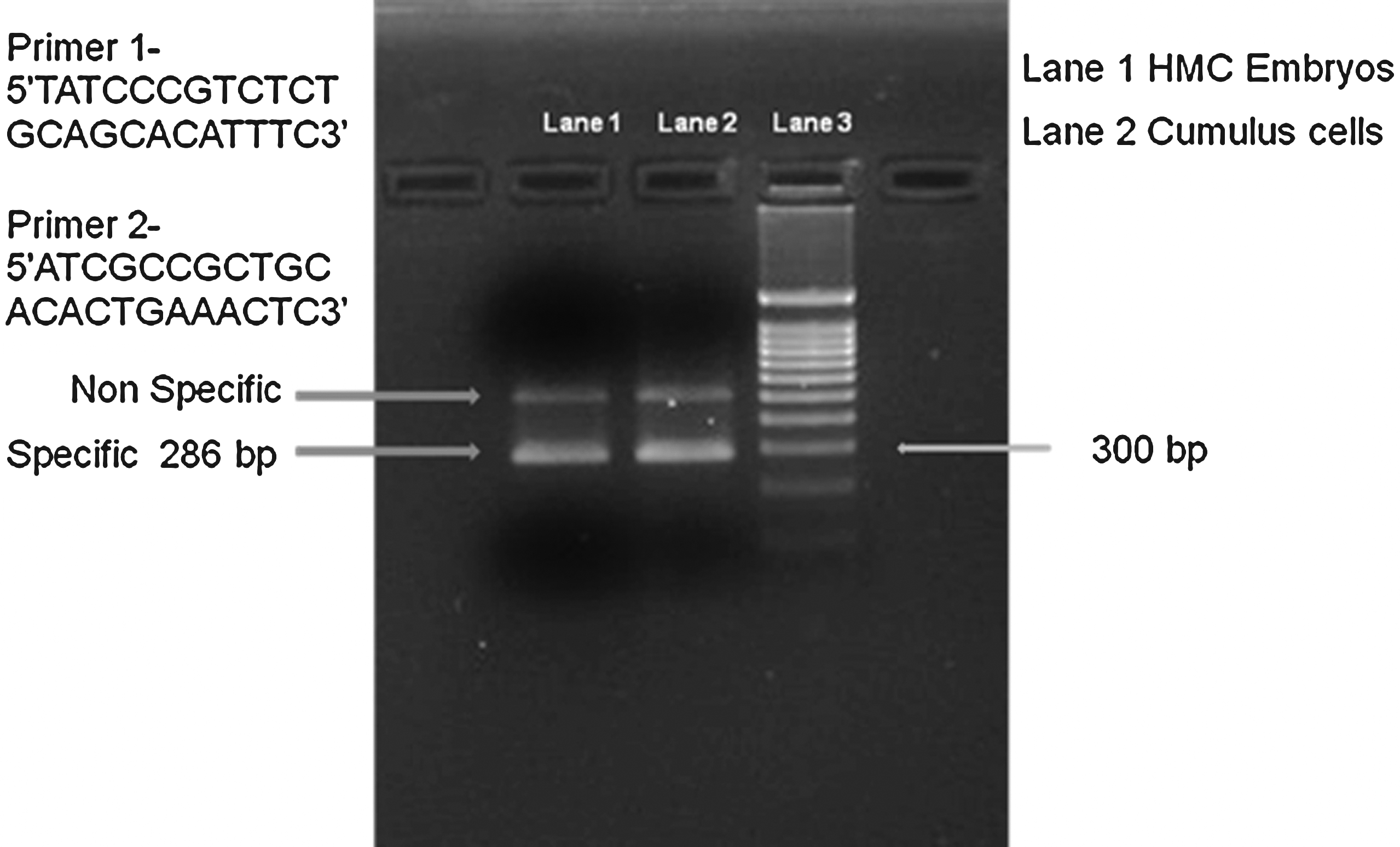
PCR analysis of the second exon of the caprine MHC class II DRB gene a. Lane 1: hand-made cloned embryos; lane 2: cumulus cells; lane 3: markers.
Discussion
In vitro cultured oocytes from different livestock species are known to take different time periods to reach the metaphase II stage. The reported time for in vitro maturation during IVF for bovine and ovine oocytes is 18–24 h (Chauhan et al., 1997a), whereas for caprine oocytes a slightly longer period of 27 h has been observed (Cognie et al., 2004; Crozet et al., 1995, Malakar and Majumdar 2005). For micromanipulator-based nuclear transfer, goat oocytes matured for 19–24 h are generally used (Daniel et al., 2008; Lan et al., 2006). In the present study as well, HMC goat oocytes were matured for 22 h before being processed for enucleation of oocytes.
For the removal of zona pellucida, several methods, viz., mechanical opening, chemical lysis, and enzymatic digestion, have been reported (Ji and Bavister 2000; Wells and Powell 2000). However, due to the slow nature of the mechanical procedure and irreversible damage caused during chemical lysis, enzymatic digestion is considered to be the preferred method for removal of zona pellucida in domestic livestock species. In the present study, enzymatic digestion using pronase was followed for zona digestion as suggested by Vajta et al. (2004). Interestingly, complete digestion of goat zona pellucida was achieved in 7 min compared to 10–15 min in cattle (Vajta et al., 2001) and 8 min in buffalo oocytes (Shah et al., 2008).
In the case of zona-digested oocytes, the first polar body moves away from the zona-free oocytes. Due to the opaqueness of the cytoplasm, the MII spindle and chromosomes remain invisible under light microscope. However, by localizing the second polar body (Pb2), the position of the chromosomes can be estimated indirectly (Fulka et al., 2001). The Pb2 remains closer to the nuclear materials because it does not have sufficient time to migrate, and thus can easily be visualized with a protrusion cone on the surface of matured oocytes. For murine and bovine oocytes, chemicals like sucrose and demecolcine (Vajta et al., 2001) are essential to make the protrusion cone clearer, whereas in the case of goat oocytes, the culture medium is found to be sufficient to produce a protrusion cone (Akshey et al., 2008).
A high rate (93%) of formation of the protrusion cone on the surface of matured oocytes was observed. In each of the oocytes, nuclear material was observed in the protrusion cone after Hoechst staining under UV light. However, damage resulting from exposure to this dye and UV light has been widely reported (Fulka et al., 2001; Galli et al., 2003). Because there is an enormous application of MII oocytes in nuclear transfer (NT), it is of immense importance to evaluate the different means to improve the enucleation process at this stage. Protrusion cone-guided bisection is considered to be one such method that can be used for enucleation of zona-free goat oocytes. In the present work, oocytes exhibiting a surface protrusion cone were selected for manual bisection. which resulted in a high (95%) enucleation rate.
The fusion efficiency of the hand-made method in cattle has been reported to be higher (90%) than the traditional NT technique (60–80%) in a number of studies (Booth et al., 2001; Kato et al., 2000; Vajta et al., 2003; Tecirlioglu et al., 2003; Wells et al., 1999). Similar observations have also been made in horses (Galli et al., 2003; Lagutina et al., 2005). In the present study, the single-step fusion (sandwich) protocol employed was observed to be more efficient (80% fusion rate) (Table 1) and convenient compared to a two-step fusion protocol. Activation of the recipient cytoplast by electric or a combination of electric and chemical stimuli (6-DMAP) is a critical element of the NT procedure. This combination has been successfully used for cattle (Cibelli et al., 1998) and rabbits (Mitalipov et al., 1999). However, prolonged exposure of 6-DMAP treatment to an electrofused oocyte–cell complex in cattle may affect the reprogramming process of the donor nuclear material resulting in chromosomal abnormalities (Van de Velde et al., 1999). On the other hand, shorter exposure of 6-DMAP does not cause any such abnormalities (Liu et al., 1998). In the case of cattle oocytes, the incubation period for activation during exposure to 6-DMAP was optimized to 4 to 6 h (Beyhan et al., 2002), whereas in the goat, it was optimized to 1–2.5 h (Lan et al., 2006). In the present study, the activation of triplets, though, was optimized to 3 h.
Chemicals like Ca ionophore and 6-DMAP have been used to obtain cloned embryos in cattle (Bhojwani et al., 2005; Tecirlioglu et al., 2005). However, in the goat, ionomycine is reported to be more effective than Ca ionophore and ethanol. Electric pulses were applied for production of cloned goat embryos using the traditional method (Melican et al., 2005; Shen et al., 2006). Melican et al. (2005) suggested that an additional electric pulse for activation was successful in producing live offspring. In the present study, electric pulse activation proved to be more efficient than Ca ionophore activation and resulted in a significant increase in cleavage and blastocyst production (data not shown).
Many cell types, including embryonic cells (Campbell et al., 1993), fibroblasts (Kato et al., 1998), mammary gland cells (Wilmut et al., 1997), cumulus cells (Wakayama et al., 1998), fibroblast cells from skin and internal organs, oviduct cells (Kato et al., 2000), sertoli cells (Ogura et al., 2000), blood leukocytes (Galli et al., 1999; Hochedlinger and Jaenisch, 2002), mural granulosa cells (Wells et al., 1999), germ cells (Bordignon et al., 2003), embryonic stem cells (Eggan et al., 2002), and colostrum-derived mammary gland epithelial cells (Kishi et al., 2000) have been successfully utilized as nucleus donors. However, it is still unclear which type cell is the most efficient for NT into oocytes (Kato et al., 2000).
Kato et al. (2000) compared efficiency of cloning using various somatic cell types from adult, newborn, and fetal female and male donor cattle. The percentage of blastocysts that developed from each of the donor cell types was not significantly different. Similarly, no differences among embryos derived from fetal and adult bovine fibroblasts with regard to fusion, cleavage, and blastocyst formation rates were detected (Nakajima et al., 2000). Similar results were obtained using various cell types derived from mice of different strains, sexes, and ages (Wakayama and Yanagimachi, 2001). In the present study, cleavage and blastocyst production rate were significantly (p < 0.05) higher when fetal fibroblast cells (92.94 ± 0.91% and 23.78 ± 3.33%) were used as nuclear donors than fresh cumulus cells (72.06 ± 2.94% and 0.00 ± 0.00%) and cultured cumulus cells (81.95 ± 3.40% and 12.74 ± 2.12%).
The embryo culture system also plays a significant role in determining the proportion of cloned embryo development to blastocysts formation. SCNT goat embryos have been cultured in 30–100-μL droplets of medium either in feeder-free culture (Das et al., 2003) or cocultured with cumulus cells (Keefer et al., 2002) with a periodic replacement with fresh medium during the 7 days of culture. The HMC embryos have been found to have a compromised development in microdrop culture (Vajta et al., 2000, 2001). Similar observations were made in present study; wherein in vitro development of HMC goat embryos in microdrops was significantly lower than the WOW and flat systems. WOW was the preferred and most efficient culture system for culturing zona-free cloned embryos (Booth et al., 2001a; Vajta et al., 2001) with a blastocyst yield of about 50% in cattle (Vajta, 2007). In comparison to cattle, lower developmental rates was observed in goats embryos in the WOW as well as the FS culture system. The FS culture system was found to be more efficient than WOW for in vitro development of HMC goat embryos. There was a significant increase in the rate of blastocyst production in flat culture than WOW (23.78 ± 3.33% vs. 15.84 ± 2.12%) Culturing of cloned embryos on a flat surface of a 4WD was advantageous, as lesser trophoblast adhesion of blastocyst to the plastic surface occurred. However, in the WOW system a higher number of embryo adhesion was observed, which has also been reported by Vajta et al. (2001). Care should be taken for successful FS culture to avoid aggregation of zona-free embryos by seeding them at a distance apart from each other, gentle handling, and undisturbed incubation until blastocyst formation.
The total number of cells (∼155) in HMC blastocysts (day 7) produced in the present study was comparable to those reported earlier in IVF goat blastocysts (data not shown). DRB gene was chosen to confirm the genetic identity of cloned goat embryos and donor cells, as they are highly polymorphic in nature. Keefer et al. (2002) has also confirmed the genetic identity of somatic cells and SCNT embryos/kids using the DRB gene.
In conclusion, a highly effective culture system for the development of HMC goat embryos has been developed, and it was observed that a flat surface system was superior in supporting the growth of HMC embryos in comparison to WOW or MD. Furthermore, fetal fibroblast cells has proved to be a better nuclear donor than fresh cumulus cells or cultured cumulus cells. This report also provides information regarding the comparison of different donor cells and culture systems in HMC embryo production in goat.
Footnotes
Acknowledgments
The authors are thankful to the Council of Scientific and Industrial Research (CSIR), India for supporting this work.
Author Disclosure Statement
The authors declare that no financial conflicts of interest exist.