
Abstract
Abstract
The birth of live animals following somatic cell nuclear transfer (SCNT) has demonstrated that oocytes can reprogram the genome of differentiated cells. However, in all species the frequency of development of healthy offspring is low; for example, in sheep, approximately only 5% of blastocysts transferred develop to term, and less than 3% develop to adulthood. Such low efficiencies, coupled with the occurrence of developmental abnormalities, have been attributed to incomplete or incorrect reprogramming. Cytoplasmic extracts from both mammalian and amphibian oocytes can alter the epigenetic state of mammalian somatic nuclei and reprogram gene expression to more resemble that of pluripotent cells. Therefore, it may be possible to increase the frequency or success of normal development by pretreating somatic cells to be used as nuclear donors prior to SCNT. In the present study, permeabilized ovine fetal fibroblasts were pretreated with a cytoplasmic extract produced from germinal vesicle (GV) stage Xenopus laevis oocytes. No increase in the frequency of development to blastocyst stage or pregnancy rate was observed; however, live birth and survival rates were significantly improved. Development to term of blastocysts transferred increased from 3.1% in the control group, to 14.7% in the treated group (a 4.7-fold increase), and even though the subsequent survival of lambs produced from treated cells was reduced by 60%, the percentage of lambs surviving to adulthood of blastocysts transferred (5.9%) increased 1.9-fold compared to controls. This study is the first to report the birth of live offspring and an increase in cloning efficiency, after crossspecies pre-reprogramming using Xenopus GV stage oocyte extract.
Introduction
Although successful across a range of species, it is generally accepted that even though differences do occur, the frequency of development to term and survival following SCNT is low in all species (Campbell et al., 2005). In addition to low efficiencies of development a range of abnormalities have been reported in SCNT-derived animals, including defects in the urogenital tract, the vascular system and the placenta (Campbell et al., 1996b; Dawson et al., 2004; Fletcher et al., 2007; Loi et al., 2006; Palmieri et al., 2007, 2008). Developmental problems associated with SCNT have been attributed to incomplete or altered reprogramming of the somatic genome (Rideout et al., 2001; Wilmut et al., 2002). In fact, it has been shown that although reprogramming in SCNT embryos is similar to the reprogramming that occurs after fertilization, these events are delayed and incomplete (Kono, 1997; Latham, 2005).
The birth of Dolly and the subsequent isolation of functional embryonic stem (ES) cells from SCNT-derived murine blastocysts provided proof that the somatic genome could be reprogrammed as the DNA beneath the epigenetic modifications remains largely unaltered (Eilertsen et al., 2007; Probst et al., 2009). As a result, a range of alternative approaches to reprogramming of somatic cells have been developed, including fusion to embryonic blastomeres (Egli et al., 2009), fusion to ES cells (Bru et al., 2008; Cowan et al., 2005; Do and Scholer, 2004; Neri et al., 2007) or embryonic germ (EG) cells (Tada et al., 1997), and exposure to extracts from somatic cells, ES cells, or mammalian (Bui et al., 2008; Miyamoto et al., 2009; Novak et al., 2004; Tang et al., 2009) or amphibian oocytes at differing stages of development (Alberio et al., 2005; Bian et al., 2009). However, as with SCNT, the extent and efficiency of complete successful reprogramming is variable and low in all cases. For example, following fusion to ES cells or treatment with ES extracts, DNA methylation status and expression of pluripotency markers Oct4, Nanog, and Rex1 are more like ES cells than the original somatic cells (Bru et al., 2008; Cowan et al., 2005; Do and Scholer, 2004; Neri et al., 2007). Despite this, the reprogramming has been shown to be only partial for fused cells and is not observed in all cells treated with ES cell extract (Collas and Gammelsaeter, 2007; Freberg et al., 2007; Neri et al., 2007; Taranger et al., 2005).
A significant advance in reprogramming of somatic cells occurred when ES-like cells (termed iPS, induced pluripotent cells) were produced by the overexpression of transcription factors; Oct3/4, Sox2, c-Myc, and Klf4. Although the efficiencies were low, this method produces either partially reprogrammed cells that activate Fbx15, an Oct4 target gene, or fully reprogrammed cells that activate the pluripotency marker genes Oct4 or Nanog (Meissner et al., 2007; Okita et al., 2007; Park and Daley, 2007; Park et al., 2008; Wernig et al., 2007).
Although the reprogramming of cells by the ex-ovo methods described above is not always complete or efficient, it has been suggested that a combination of reprogramming techniques may improve the efficiency or frequency of normal development of SCNT-derived animals (Campbell and Alberio, 2003; Gurdon and Murdoch, 2008). Indeed, pretreatment of murine fibroblasts with murine GV stage oocyte extract before SCNT resulted in a 3.4-fold increase in the production of cloned offspring, compared with SCNT controls (Bui et al., 2008). Here we hypothesize that treatment of somatic cells with extracts prepared from GV stage Xenopus laevis oocytes prior to their use as nuclear donors for SCNT will improve subsequent development. GV stage Xenopus oocytes were used as they provide an abundant source of material, and previous studies using GV stage extracts from various species have shown them to remodel epigenetic markers associated with nuclear and chromatin structure (Alberio et al., 2005; Bian et al., 2009) and directly activate stem cell marker genes (Bui et al., 2008; Miyamoto et al., 2007, 2008, 2009; Tang et al., 2009) without cell division or protein synthesis (Gurdon, 1999). In these studies the effects of pretreatment of ovine fibroblasts with Xenopus (GV stage) oocyte extract, before SCNT on embryo development, establishment of pregnancy, birth of live offspring, survival, and occurrence of developmental abnormalities was determined.
Materials and Methods
All chemicals were purchased from Sigma-Aldrich Company (St. Louis, MO, USA) unless otherwise stated.
All experiments involving animals were performed under Home Office Regulation.
Collection and in vitro maturation of ovine oocytes
Sheep ovaries were collected at a local slaughterhouse and transported to the laboratory in phosphate-buffered saline (PBS) at 25°C. Antral follicles, 2–3 mm diameter, were aspirated using a 10-mL syringe fitted with a 28-G needle; only good-quality oocytes surrounded by at least three layers of cumulus cells and exhibiting a homogeneous cytoplasm were selected for in vitro maturation. Selected oocytes were washed three times in HEPES-buffered TCM 199 (Gibco, Paisley, UK) supplemented with 10% fetal bovine serum (FBS) at 39°C. For maturation 45–50 oocytes were cultured in maturation medium [bicarbonate-buffered TCM 199 (Gibco) supplemented with 10% FBS, 5 μg/mL follicle stimulating hormone (Vetropharm, Belleville, Canada), 5 μg/mL Luteinizing hormone (Vetropharm), 1 μg/mL estradiol-17β, 0.3 mM sodium pyruvate, 100 μM cysteamine, and 50 μg/mL gentamycin] under mineral oil in four-well dishes (Nunc, Naperville, IL, USA) at 39°C, in a humidified atmosphere of 5% CO2.
Donor cell culture
Ovine primary fetal fibroblasts were isolated as previously described (Wilmut et al., 1997) from a day 30 fetus obtained from a purebred Llynn ewe and cultured for two passages in Dulbecco's modified eagle's medium (DMEM), supplemented with 1% MEM-nonessential amino acids, 1 unit/mL penicillin, 0.1 mg/mL streptomycin, and 10% FBS. Primary cultures were then stored in liquid nitrogen until required. For each experiment, cells were thawed and cultured until 80–90% confluent; quiescence was then induced by reducing the concentration of FBS in the supplemented DMEM to 0.1% for 5 or 7 days.
Preparation of Xenopus oocyte extract
Mature female Xenopus laevis were euthanized by submersion in MS 222, the ovaries were removed and dissected into small pieces, and placed into 25-cm Petri dishes containing 30–40 mL collagenase solution (8 mg/mL type II collagenase in calcium free Ringers solution) for 2–3 h, on a rocking table at room temperature (RT). After complete digestion, free oocytes were removed and washed five times in excess 0.9% saline followed by three washes in excess ice-cold extraction buffer (20 mM HEPES, 100 mM potassium chloride, 5 mM magnesium chloride, 2 mM β-mercaptoethanol, 6.3 μM leupeptin, 0.15 μM aprotinin, and 1.5 μM pepstatin A). Oocytes were transferred into 15 mL polypropylene centrifuge tubes with a round bottom (Greiner) and allowed to settle under gravity, excess buffer was then removed and the oocytes centrifuged at 10,000 × g for 10 min at 4°C. The middle ooplasmic layer was removed and recentrifuged for a further 10 min to remove debris. The supernatant was collected, transferred into 5-mL polyallomer tubes (Beckman, Palo Alto, CA, USA), and centrifuged at 100,000 × g for 40 min at 4°C using an ultracentrifuge (L8-70M; Beckman). The supernatant was transferred into clean tubes and then recentrifuged for 30 min. Glycerol was added to the oocyte extract to give a final concentration of 5% (v/v), and the extract then aliquoted into 0.5 mL aliquots, snap frozen in liquid nitrogen, and then stored at −80°C.
Incubation of somatic cells in Xenopus oocyte extract
Quiescent donor cells were harvested by trypsinization, washed in permeabilization buffer (170 mM potassium gluconate, 5 mM potassium chloride, 2 mM magnesium chloride, 1 mM potassium phosphate, 1 mM EGTA, and 20 mM HEPES, pH 7.25, and 2 mM dithiothreitol, 6.3 μM leupeptin, 0.15 μM aprotinin, and 1.5 μM pepstatin A, with an osmolarity of 352 mOsm) and then permeabilized by suspension in digitonin solution (20 μg/mL digitonin in permeabilization buffer) for 2 min on ice, at a concentration of 2 million cells per 1 mL. Permeabilization was stopped by adding an excess of permeabilization buffer and centrifuging at 700 × g for 10 min. The percentage of cells permeabilized was estimated using FITC-labeled dextran; a 5-μL aliquot of permeabilized cells was added to 10 μL of 5 mg/mL FITC-labeled dextran in PBS, the mixture was pipetted onto a glass slide, covered with a glass coverslip, and imaged using a fluorescent microscope (Leica, Deerfield, IL, USA). Permeabilized cells were incubated in either supplemented DMEM (containing 10% FBS) for controls or freshly thawed Xenopus oocyte extract, at a concentration of 3000 cells/μL for 3 h at 17°C, which was based on previous studies (Alberio et al., 2005) and previous in-house data (personal communication with S. Lazar). After incubation cells were washed by centrifugation in permeabilization buffer, and an aliquot of the cells taken and spun onto coverslips for immunocytochemistry, using a cytospin centrifuge (Centurion, 2000 series). The remaining cells were resuspended in starvation media (supplemented DMEM with 0.1% FBS) and were used for SCNT. The production of control (permeabilized) cells and extract treated cells was undertaken once a week and this was randomized each week, this gives five experimental data sets (each containing 4–5 surrogate ewes/replicates) for each group.
Somatic cell nuclear transfer (SCNT)
All procedures were carried out as previously described (Lee and Campbell, 2006). Briefly, at 15 h postonset of maturation (hpa), oocytes were stripped of cumulus cells by vortexing for 2–3 min in HEPES-buffered Synthetic Oviduct Fluid (H-SOF) medium containing 300 IU/mL of hyaluronidase, and then washed three times in H-SOF. Enucleation was carried out by removing a portion of cytoplasm containing the extruding anaphase/telophase of the first meiotic division (AI-TI) spindle, using a 20-μm glass micropipette, in H-SOF containing 4 mg/mL bovine serum albumin (BSA) and 15.6 μM cytochalasin B (CB). Enucleated oocytes were returned to culture in maturation medium and then transferred to maturation medium supplemented with 10 mM caffeine for 6 h from 18–24 hpa. At 24 hpa a single permeabilized ovine fetal fibroblast, previously incubated in supplemented DMEM or Xenopus oocyte extract, was fused with an enucleated cytoplast using two DC pulses of 1.25 kV/cm for 30 μsec in 0.3 M mannitol in the absence of calcium, using an Eppendorf Multiporator (Eppendorf, Germany). Couplets were returned to maturation medium and at 25 hpa, fused couplets were selected and activated by a 5-min exposure to 5 μM calcium ionophore followed by incubation in modified SOF (mSOFaciBSA) medium, containing 2% BME-essential amino acids, 1% MEM-nonessential amino acids, and 4 mg/mL BSA, supplemented with 35.5 μM cycloheximide and 15.6 μM CB for 5 h at 39°C, in a humidified atmosphere of 5% CO2. After activation, presumptive embryos were cultured in 50 μL drops of mSOFaciBSA, under mineral oil at 39°C in a humidified atmosphere of 5% CO2 and 5% O2. Cleavage was assessed on day 2 of culture and embryo development on day 7 when blastocyst stage embryos were surgically transferred into the uterus of synchronized surrogate ewes. One to three blastocysts were transferred per recipient, and four or five ewes were used per experimental set.
Immunocytochemistry for HistoneH3K9 and DNA methylation
For staining of histone H3 trimethylated at K9 (TriH3K9Me); cells attached to coverslips were fixed in ice-cold methanol at −20°C for 20 min, washed three times for 5 min in PBS-Tween (PBS, 0.1% Tween 20, and 7.7 mM sodium azide) and treated with 0.5% Triton X-100 in PBS at RT for 30 min. The cells were then washed in PBS-Tween, prior to incubation in blocking solution (5% BSA in PBS-Tween) for 60 min at RT. Cells were then incubated in primary antibody rabbit antimammal primary antibody (Abcam) diluted 1:500 in blocking buffer, for 2 h at RT. The cells were washed three times for 5 min in PBS-Tween and incubated with the secondary antibody; FITC-conjugated donkey antirabbit (Dako, Carpinteria, CA, USA) diluted 1:200 in blocking buffer, for 1 h at RT. The cells were washed three times for 5 min in PBS-Tween and mounted onto slides using Vectorshield containing DAPI (Vector Laboratories Inc., Burlingame, CA, USA), sealed with nail varnish, and stored at −20°C until imaging. For 5-methylcytosine (5MeC) staining the method was the same as for TriH3K9Me, except cells were fixed using 1.3 M paraformaldehyde for 15 min at RT, the cells were incubated in 4 M hydrochloric acid for 30 min at RT instead of Triton X-100, the primary antibody was a mouse antivertebrate (Abcam) diluted 1:500, the secondary antibody was a FITC-conjugated rabbit antimouse (Dako) diluted 1:20, after the last wash the cells were incubated for 15 min at RT in 37 μM propidium iodide (PI) in PBS, then washed, mounted in vectorshield, sealed, and stored at −20°C.
Slides were examined using a fluorescent microscope (Leica) using 100 × magnification. Quantitative analysis was undertaken using SIMPLE PCI software (Compix Inc., Lake Oswego, OR, USA), and methylation status calculated as the intensity of FITC staining divided by the intensity of nuclear staining with DAPI/PI.
Statistical analysis
The t-tests were carried out using Prism (GraphPad Software, Inc., La Jolla, CA, USA), and GenStat (VSN International Ltd, Hemel Hempstead, UK) was used for all other analyses. Results were deemed statistically significant over a 95% confidence interval.
Results
Effect of permeabilization and Xenopus oocyte extract on donor cells
The percentage of cells recovered after permeabilization was on average 75% (±5% SEM) and of these cells approximately 80% (±5% SEM) were permeabilized (n = 18).
After incubation in the extract/supplemented DMEM all cells were used for SCNT. Some of the cells remaining after SCNT were plated for culture in supplemented DMEM and their recovery noted; both control and extract treated cells contained vacuole-like structures for up to 7 days (see Supplementary Fig. 1; see online supplementary material at www.liebertonline.com). After 7 days, recovering cells displayed normal fibroblast morphology.
Effects of Xenopus oocyte extract on HistoneH3K9 and DNA methylation in donor cells
DNA methylation status was determined using an antibody to 5MeC; a decrease in 5MeC is indicative of demethylation at CpG dinucleotides, the most common areas on the DNA of mammals that methyl groups bind (Jabbari and Bernardi, 2004; Spivakov and Fisher, 2007). The methylation status of H3K9 was demonstrated using an antibody specific to the trimethylated form of H3K9 (TriH3K9Me); the global loss of methyl groups at the lysine base to form dimethylated or methylated H3K9 has been implemented as a marker of ES cells (Atkinson and Armstrong, 2008).
The intensity of TriH3K9Me staining in the cells was significantly reduced when treated with the Xenopus oocyte extract (control 1.05 ± 0.04, treated 0.76 ± 0.03, p < 0.0001; two-tailed unpaired Welch's t-test) (Fig. 1a and b). However, the intensity of 5MeC staining in the cells was not significantly reduced after treatment, although a reduction in intensity was observed (control 1.87 ± 0.09, treated 1.66 ± 0.09, p = 0.11; two-tailed unpaired Welch's t-test).
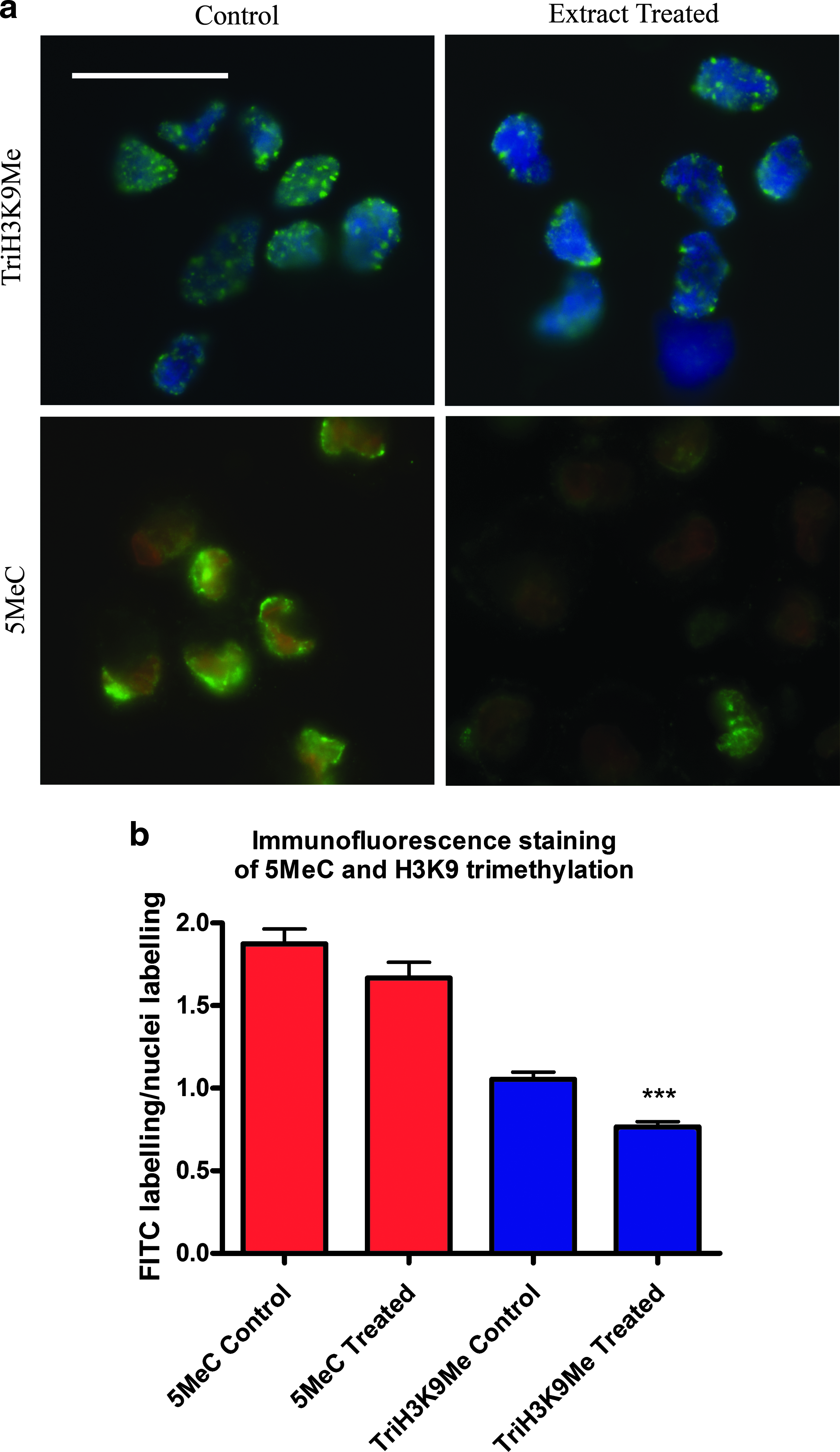
Quantitative analysis of the methylation status of 5MeC and TriH3K9Me in control and Xenopus oocyte treated cells using immunostaining. The level of methylation was detected using FITC-labelled antibodies bound to antibodies specific to 5MeC and TriH3K9Me. (
Fusion and development of SCNT ovine embryos
No difference was observed in the number of fused couplets or the number of fused couplets that cleaved between the extract treated or control groups (p = 0.89 for both; two-tailed unpaired Welch's t-test). There was a small increase in the percentage of fused couplets that developed to the blastocyst stage in the Xenopus oocyte extract treated group (Table 1), however, this was not significantly significant (p = 0.58; two-tailed unpaired Welch's t-test).
Pregnancy and birth rates of SCNT-derived blastocyst stage embryos transferred to surrogate recipients
The number of recipient ewes pregnant at 6 weeks was the same for both the control and Xenopus oocyte extract treated groups, and at 12 weeks there was only one additional ewe pregnant in the extract treated group. Therefore, there was no significant difference in the pregnancy rates between the two groups at either scan time point. However, there were significant differences in the live birth rates between the control and extract groups, with regard to pregnancies detected at each scan time point (p = 0.013 and p = 0.008 for the 6 and 12 week scans, respectively, using chi-square test). The number of live births from each group with regard to the number of recipients that were pregnant at the second scan showed a dramatic increase, from 25% in the control group to 88.9% in the extract treated group (Table 2). It was also noted that in the third trimester of pregnancy three of the pregnant recipient ewes in the control group aborted, compared to only one in the extract treated group. The number of live offspring from each group with regard to the number of blastocysts transferred into each recipient ewe (total of 48 experimental recipients, 24 per group) was analyzed using a generalized linear model assuming binomial errors. There was no evidence for a systematic difference between the five sets within each group (p = 0.634; analysis of deviance), and the evidence for the extract treated group's sets producing more lambs was significant (p = 0.011; analysis of deviance). With the sets omitted from the analysis, subsequent analysis of the individual replicates (recipient ewes) showed the evidence for the extract treated group producing more live lambs to be even more significant (p = 0.009, using analysis of deviance). The predicted proportions of lambs from embryos for each group were; control group, 3.1% (±1.9) and the extract treated group, 14.7% (±3.8) (Table 2).
Superscripts within columns represent statistical difference. a–b(p = 0.008), c–d(p = 0.009)
Six of the 10 lambs died or were terminated shortly after birth.
In the Xenopus oocyte extract treated group 6 of the 10 lambs died or were terminated shortly after birth (5 lambs at 0–8 days and 1 lamb at 40 days). Post mortems of the lambs revealed kidney and/or heart abnormalities in the majority of cases (including large aorta and grossly enlarged renal pelvises). The surviving lambs in both groups are healthy and are now greater than 18 months old.
Discussion
GV and MII stage oocyte cytoplasm from both amphibia and mammals have shown differences in their reprogramming capabilities (Alberio et al., 2005; Bian et al., 2009; Bui et al., 2008), although due to technical difficulties in mammals live offspring have only been generated using MII oocytes as cytoplast recipients for SCNT (Campbell et al., 1996c). Mammalian somatic cells do not undergo DNA replication during incubation in GV stage oocyte extract, and afterward 99% show Lamin A/C removal (Alberio et al., 2005), suggesting considerable nuclear remodeling. When these treated cells are cultured they form ES cell-like colonies and show activation of pluripotent marker genes after 7 days, in particular, NANOG (Miyamoto et al., 2009). Somatic cells injected into GV oocytes have been shown to be completely demethylated at H3K9 after 7 h, H3K9 methylation is normally epigenetically stable in somatic cells and even in cells injected into MII oocytes (Bui et al., 2008). MII oocyte extracts have been shown to support DNA replication, remove the TATA binding protein (transcription factor that binds to the TATA box region) and Lamin A/C, but to a lesser extent (74 %) than GV stage extract, as well as erase the machinery for regulating gene expression in somatic cells (Alberio et al., 2005; Miyamoto et al., 2009). However, these cells did not show any obvious changes in pluripotency marker genes or the formation of colonies, after cell culture (Miyamoto et al., 2009). These observations indicate that GV stage oocyte cytoplasm could be superior to MII stage in its reprogramming capabilities.
Research by Tang et al. (2009) has shown that bovine donor cells pretreated with bovine MII stage extract increased blastocyst development when used as nuclear donors for SCNT, whereas no increase was observed in GV stage oocyte extract treated cells over nontreated controls (Table 3). This study supports the observations of Tang et al. (2009) in that the pretreatment of the donor cells with Xenopus GV extract did not improve the development to blastocyst of reconstructed embryos (Table 3).
ND, not determined.
Efficiency of cloning by SCNT is known to be poor in sheep with approximately only 5% of transferred cloned embryos developing to term (Campbell et al., 1996d; Rideout et al., 2001; Wilmut et al., 1997, 2002). An increase in cloning efficiency with Xenopus GV stage oocyte extract treatment has been shown in this study; by an increase in the number of blastocysts developing into live offspring, from 3.1% in the control group (permeabilized cells) to 14.7% (a 4.7-fold increase). Typically, across species less than 3% of embryos derived by SCNT develop into adult animals (Rideout et al., 2001; Wilmut et al., 2002), which is believed to be due to insufficient reprogramming. This percentage is very similar to our control group (using permeabilized cells), where only 3.1% of the blastocysts transferred into recipient ewes developed into adult animals. In this study we also show that the pretreatment of donor cells with Xenopus oocyte extract increased the percentage of animals surviving to adulthood to 5.9% (a 1.9-fold increase from the permeabilized controls), even with a 60% loss of lambs from the treated group. The results described in this study show a similar trend to work reported by Bui et al. (2008), who used murine somatic cells prereprogrammed with murine GV stage oocyte extract; they reported that these extract-treated SCNT-derived embryos showed an 3.4-fold increase in the production of live offspring when compared to intact SCNT controls, and a 7.7-fold increase when compared to permeabilized SCNT controls (Table 3). Research has also shown that bovine oocyte cytoplasm can support the development of SCNT embryos derived from a variety of different mammalian donor cells, and suggests that the mechanisms regulating early embryonic development maybe conserved among mammals (Dominko et al., 1999). Nevertheless, this study is the first to report live offspring and an increase in the efficiency of cloning, after cross species prereprogramming using Xenopus laevis GV stage oocyte extract, and supports the theory that mechanisms regulating early embryonic development are conserved further down the vertebrate evolutionary tree (Bian et al., 2009).
Footnotes
Acknowledgments
We thank David Edwards, Michael Baker, Debbie Surgay, Morag Hunter, and staff of the Biosciences Resource Unit for assistance with embryo transfer, animal husbandry, and ultrasound scanning. This work was supported by the European Science Foundation (EuroSTELLS), DTI and CellCentric (Cambridge).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.