
Abstract
Abstract
Human-induced pluripotent stem cells (iPSCs) generated from human adult somatic cells through reprogramming hold great promises for future regenerative medicine. However, exposure of human iPSCs to animal feeder and serum in the process of their generation and maintenance imposes risk of transmitting animal pathogens to human subjects, thus hindering the potential therapeutic applications. Here, we report the successful generation of human iPSCs in a feeder-independent culture system with defined factors. Two stable human iPSC lines were established from primary human dermal fibroblasts of two healthy volunteers. These human iPSCs expressed a panel of pluripotency markers including stage-specific embryonic antigen (SSEA)-4, tumor-rejection antigen (TRA)-1-60, TRA-1-81, and alkaline phosphatase, while maintaining normal karyotypes and the exogenous reprogramming factors being silenced. In addition, these human iPSCs can differentiate along lineages representative of the three embryonic germ layers upon formation of embryoid bodies, indicating their pluripotency. Furthermore, subcutaneous transplantation of these cells into immunodeficient mice resulted in teratoma formation in 6 to 8 weeks. Our findings are an important step toward generating patient-specific iPSCs in a more clinically compliant manner by eliminating the need of animal feeder cells and animal serum.
Introduction
Methods
Derivation of primary human dermal fibroblasts
Our study protocol for the procurement of human tissue for use in reprogramming experiments was approved by the institutional review board of Queen Mary Hospital, Hong Kong, and was registered at the Clinical Trial Center, the University of Hong Kong, number HKCTR-725 (http://www.hkclinicaltrials.com). Voluntary prior informed consents (PIC) were obtained from all participants. Using an aseptic technique, skin biopsies were obtained from the forearms of two healthy volunteer Chinese men (37 and 57 years old) with 6-mm punch biopsy needles. The biopsies were minced and plated in six-well plates with mTeSR™1 medium (StemCell Technologies Inc.). After 7 to 14 days, dermal fibroblasts grew out from the skin tissues, and were trypsinized for further expansion.
Lentiviral production
The lentiviral plasmids pSin-hOCT4 (NM 002701), pSin-hLin28 (NM024674), pSin-hSox2 (NM003106), and pSin-hNanog (NM024865) were obtained from Addgene Inc. (Cambridge, MA, USA) (www.addgene.org). hKLF-pCMV-Sport6 (NM004235) and hc-Myc-pCMV-Sport6 (NM002467) were obtained from Open Biosystems (Australia) (www.openbiosystems.com) and subcloned into pSin plasmid from Addgene Inc. In addition, lentiviral-green fluorescent protein (GFP) under EF1α promoter was used for control experiment. These lentiviruses were packaged with lipofectamine™ 2000 (Invitrogen) in the presence of lentiviral vectors (pCMV-dR8.91 and pMDLg/pRRE) (Tronolab, Lausanne, Switzerland) using 293FT cells (Invitrogen). The resultant viral supernatant was harvested and concentrated by standard protocol. Viral titers were determined by Lenti-X™ qRT-PCR Titration Kit (Clontech Laboratories Inc., Mountain View, CA, USA), and the multiplicity of infection was 10.
Generation of human iPSCs in feeder-independent system
Dermal fibroblasts were plated in six-well plates at density of 1 × 105 cells per well, and were transduced with 100 μL of each virus together with 6 μg/mL polybrene (Sigma Aldrich, St. Louis, MO, USA) at 37°C for 24 h. The transduced cells were then trypsinized and replated onto Matrigel™ (1:30, BD Biosciences, San Jose, CA, USA)-coated six-well plates. Two days after transduction, the mTeSR™1 medium was supplemented with 50 ng/mL bFGF (PeProtech Inc., Rockhill, NJ, USA). Seven days after transduction, 10 μM of Y-27632 (CalBiochem, EMD Chemicals Inc., Darmstadt, Germany), a selective Rho-associated kinase (ROCK) inhibitor, was added for 24 h. Putative human iPSC colonies were manually dissected and subcultured onto new Matrigel™-coated wells for the first five passages. Thereafter, colonies were detached with 1 mg/mL dispase, then cut evenly by pasture pipette and subcultured.
Flow cytometry
Reprogrammed iPSC colonies were harvested as single cells by TripLE solution digestion (Invitrogen). Cells were then fixed and permeabilized in 2% paraformaldehyde with 0.05% Triton X-100. Cells were stained by rabbit or mouse antihuman Oct-4, Nanog, SSEA-4, TRA-1-60 antibodies (Stemgent, San Diego, CA, USA) and TRA-1-81 (BD Biosciences) and analyzed on a FC500 flow cytometer (Beckman Coulter, Fullerton, CA, USA) using CXP analysis software (Beckman Coulter). Rabbit or mouse isotypic antibodies were used as a negative control.
Karyotype analysis
Cells at the 10th to 12th passage were blocked at metaphase by exposing to colcemid (0.025 μ/mL; GIBCO, Invitrogen Corp., Paisley, UK) for 14 h. After hypotonic and fixative treatment, the cell suspension was spread on slides, and chromosomal analysis was performed on Giemsa-banded metaphases using a bright-field microscope.
Alkaline phosphatase and Immunofluorescence staining
Alkaline phosphatase staining was preformed according to the manufacturer's protocol (Stemgent). For immunofluorescence staining, colonies were fixed and permeabilized in 2% paraformaldehyde with 0.05% Triton X-100 solution and subsequently stained with pluripotency markers (antihuman SSEA-4, TRA-1-60, Oct-4, and Nanog) (Stemgent). Positive colonies were visualized by adding rabbit antimouse IgG H+L Alexa 488 and goat antirabbit IgG H+L Alexa 594 (Molecular Probes, Eugene, OR, USA) for 30 min. Images were acquired using Ziess fluorescence microscope with AxioVision software (Ziess GmbH, Gottingen, Germany).
Reverse-transcription polymerase chain reaction (PCR)
Total RNA from cells at various stages was extracted with TRI® reagent (Applied Biosystems, Bedford, MA, USA). Reverse transcription was performed with QuantiTect® reverse transcription kit (Qiagen, Hilden, Germany) according to the manufacturer's instruction. PCR was performed with Ampli Gold Taq Polymerase and GenAmp PCR system 9700 (Applied Biosystems). Primers sequence, annealing temperatures, and product size were depicted in Table 1. GAPDH was used as an internal control for RT-efficiency and RNA integrity.
REX1, RNA exonuclease 1; ICAM, intercellular adhesion molecule 1; IHH, Indian hedgehog homolog; AFP, alpha-fetoprotein; GAPDH, glyceraldehde 3-phosphate dehydrogenase (Xu et al., 2001).
Bisulfite Pyrosequencing
Genomic DNA (1 mg) was bi-sulphite-treated using EZ DNA methylation kit (Zymo Research, Orange, CA, USA), according to the manufacturer's instructions. The promoter regions of the human Oct3/4 gene was amplified using Amplitaq gold 360 (Applied Biosystems) with a biotinylated reverse primers. The PCR products were sequenced by Pyrosequencing PSQ96 HS System (Biotage, Uppsala, Sweden) according to the manufacturer's instructions. The methylation status of each locus was analyzed using Pyro Q-CpG software (Qiagen).
In vitro differentiation
For embryoid body formation, putative human iPSC colonies were first dissociated to single cells with TripLE solution (Invitrogen). Hanging drops of 1000 cells in 20 μL culture medium in the absence of bFGF were then formed onto the lids of a Petri dish for 48 h. Thereafter, embryoid bodies were suspended in DMEM supplemented with 10% FBS, 1× NEAA and 1 mM L-glutamine for 5 days, followed by plating onto gelatin-coated 12-well plates for another 7 days in the same culture medium. The embryoid bodies were then fixed and stained with human embryonic germ layer characterization kit (Chemicon, Temecula, CA, USA).
For endothelial progenitor cell (EPC) differentiation, embryoid bodies obtained after suspension were plated in gelatin-coated dishes for 5 days, central regions were dissected, and cultured in EGM-2 medium for 14 days (Lonza Inc., Walkerville, MD, USA). CD34-positive cells were then sorted by Microbeads cell sorting kit (Miltienyi Biotech GmBH, Bergish Gladbach, Germany). For EPC characterization, cells were treated with 4.8 μg/mL of DiI-labeled acetylated low-density lipoprotein (DiI-AcLDL, Molecular Probes), followed by fixation in 2% paraformaldehyde, and immunostaining with 10 μg/mL of Ulex europaeus Lectin-FITC (Sigma-Aldrich, St. Louis, MO, USA).
Angiogenic tube formation assay
Tube formation of the human iPSC-derived EPCs was preformed by the In Vitro Angiogenesis Assay Kit (Chemicon). Briefly, harvested EPCs were resuspended in EGM medium, 10,000 cells were seeded per well in ECMatrix™ solution precoated 96-well plate and incubated at 37°C for 6 h. Mesh-like structures were observed in cells with endothelial properties under an inverted light microscope.
Migration assay
Human iPSC-derived EPCs were placed in the upper chamber of the Transwell® pore Polycarbonate Membrane Insert (Corning, Lowell, MA, USA). The chamber was placed in a 24-well plate containing EBM medium with 50 ng/mL VEGF (PeProtech Inc.) and incubated at 37°C for 24 h. Then the lower side of the filter was washed with phosphate-buffered saline (PBS) and fixed with 2% paraformaldehyde. Migrated cells were visualized by hematoxylin and eosin staining.
Teratoma formation
Human iPSCs were injected subcutaneously into nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice (1 × 106 cells per site). After 6 to 8 weeks, tumors were resected, fixed in 4% buffered formalin, and then processed for paraffin sectioning. Immunohistological staining of the three germ layers was then preformed using the human embryonic germ layer characterization kit (Chemicon).
Statistical analysis
Continuous variables are expressed as mean ± SEM. Statistical comparisons were performed using Student's t-test. Calculations were performed with GraphPad Prism 5 (GraphPad, La Jolla, CA, USA). A p-value <0.05 was considered statistically significant.
Results
Generation of human iPSCs in feeder-independent system with defined factors
We first attempted to generate human iPSCs using a feeder-independent culture system with Matrigel™ as culture matrix and mTeSR™1 stem cell medium. In the initial series of experiments, primary human dermal fibroblasts obtained from skin biopsies of two healthy Chinese men, were expanded in mTeSR™1 stem cell medium, followed by lentiviral transduction of six reprogramming transgenes (Oct4, Sox2, Klf4, c-Myc, Lin28, and Nanog). Ten days posttransduction, clusters of round cells with a loose and granular morphology started to appear, whereas no such clusters were observed in the control experiments (Fig. 1G–H). However, these clusters failed to expand and stopped proliferating 28 days posttransduction, despite successful viral transduction (Fig. 1A–F).

Initial trials of human iPSC generation in feeder-independent, serum-free condition.
Previous works have shown that Rho-associated kinase (ROCK) inhibition can improve dissociation-induced apoptosis, and apoptosis in serum-free suspension of human ESCs as well as human iPSCs (Mollamohammadi et al., 2009; Watanabe et al., 2007). As a result, we tried to improve the reprogramming process by the application of a selective ROCK inhibitor, Y-27632 (10 μM) to the cells at posttransduction day 7 for 1 day. The protocol was summarized in Figure 2A. Approximately 14 days after viral transduction, small compact clusters (10–20 clusters per well) of cells with morphology resembling to that of human ESCs with high nucleus-to-cytoplasm ratio, prominent nucleoli, and pronounced individual cell borders were observed (Fig. 2B). These putative human iPSC clusters were handpicked to subculture to new Matrigel™-coated wells in mTeSR™1 medium (Fig. 2C–F). They progressively increased in size with strong expression of alkaline phosphatase (Fig. 2G). To quantify the effects of ROCK inhibition on human iPSC generation, primary human dermal fibroblasts isolated from two healthy Chinese males (KS1 and KS2) were reprogrammed according to the protocol depicted in Figure 2A with and without Y-27632 on day 7 for 1 day, and the number of ESC-like cell clusters per 10,000 cells were counted on day 14. Figure 3 showed that although ESC-like cell clusters were observed in cells reprogrammed with and without ROCK inhibition, the number of ESC-like cell clusters obtained at day 14, cells reprogrammed with ROCK inhibition had a significantly higher number of ESC-like clusters than the counterpart without ROCK inhibition (12.4 ± 1.93 ESC-like clusters/10,000 cells vs. 2.8 ± 0.66 ESC-like clusters/10,000 cells, p < 0.05). In fact, no ESC-like clusters survived to day 28 in the absence of ROCK inhibition (Fig. 3). In addition, primary adult dermal fibroblasts treated with Y-27632 in the same culture condition but without lentiviral transduction did not yield any reprogrammed colonies (data not shown).

Generation of human iPSCs with feeder-independent, serum-free condition with defined medium.
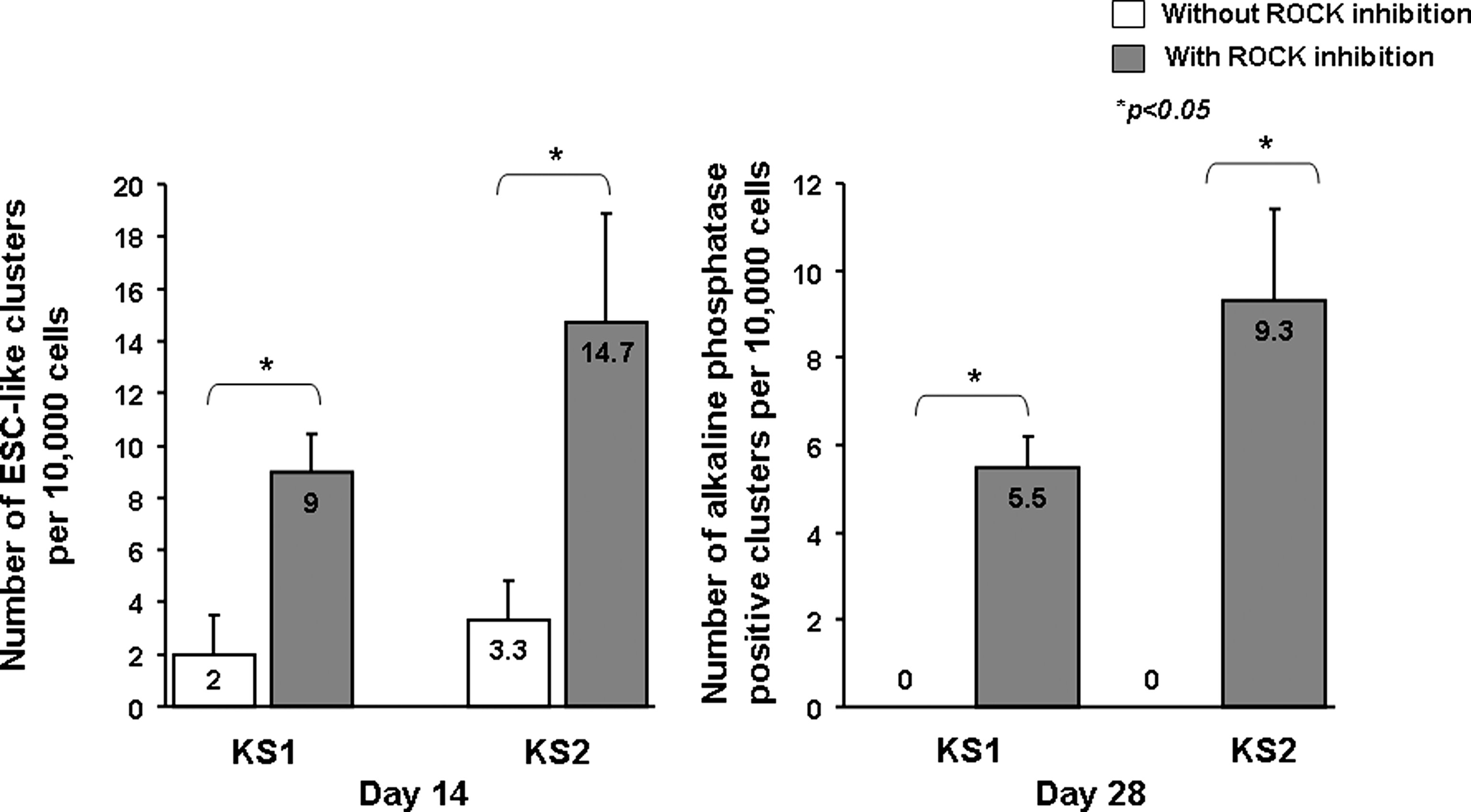
The number of ESC-like clusters 14 days and 28 days after reprogramming of primary human dermal fibroblasts obtained from two healthy Chinese males with and without ROCK inhibition (n = 3).
Pluripotency markers
We then examined whether these putative human iPSCs exhibit hESC-like properties. Immunofluorescence revealed expressions of a panel of pluripotency markers including Oct4, Nanog, KLF, SSEA-4, TRA-1-60, and SOX2 (Fig. 4A–B) in the two putative human iPSC lines, KS1-iPSC and KS2-iPSC, resembling that of human embryonic stem cell line H9 (as control; Fig. 4C). Consistently, flow cytometry analysis showed that the putative human iPSCs were positive for Oct-4 (98.5 ± 1.8%) SSEA-4 (99.1 ± 1.1%), TRA-1-60 (93.8 ± 3.6%), and TRA-1-81 (99.7% ± 0.1%) (Fig. 4D) comparable to that of hESC lines H7 and H9. In addition, both KS1-iPSC and KS2-iPSC lines maintained normal karyotypes as revealed by cytogenetic analysis (Fig. 5A). To further demonstrate the epigenetic reprogramming, we analyzed the methylation status of the Oct4 promoter region of the putative human iPSC lines with quantitative bisulphate pyro-sequencing. Although the Oct4 promoters of the parent fibroblasts (KS1 and KS2) remained heavily methylated, extensive demethylation was observed in both KS1-iPSC and KS2-iPSC, similar to that of hESC-H9, representing the reactivation of the key pluripotency gene (Fig. 5B). Furthermore, qPCR analysis revealed the silencing of expressions of exogenous transgenes, OCT4 and Nanog, and the upregulation of the endogenous Oct4 and Nanog (Fig. 5C). At the time of the writing of this manuscript, they were propagated over 20 passages.

Characterization of the human iPSC lines. Immunofluorescence analysis of human iPSC lines

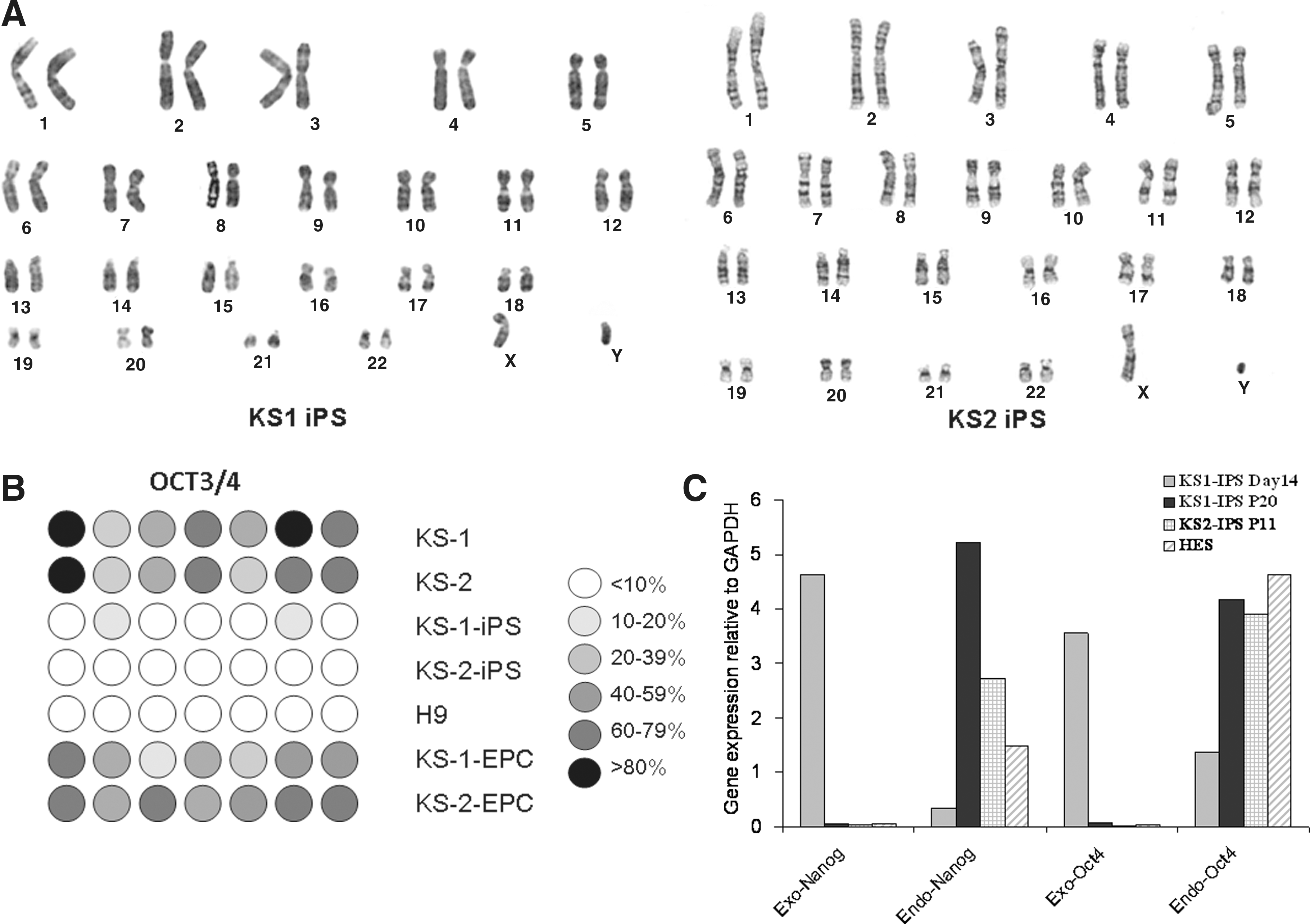
In vitro and in vivo pluripotency tests of putative human iPSCs
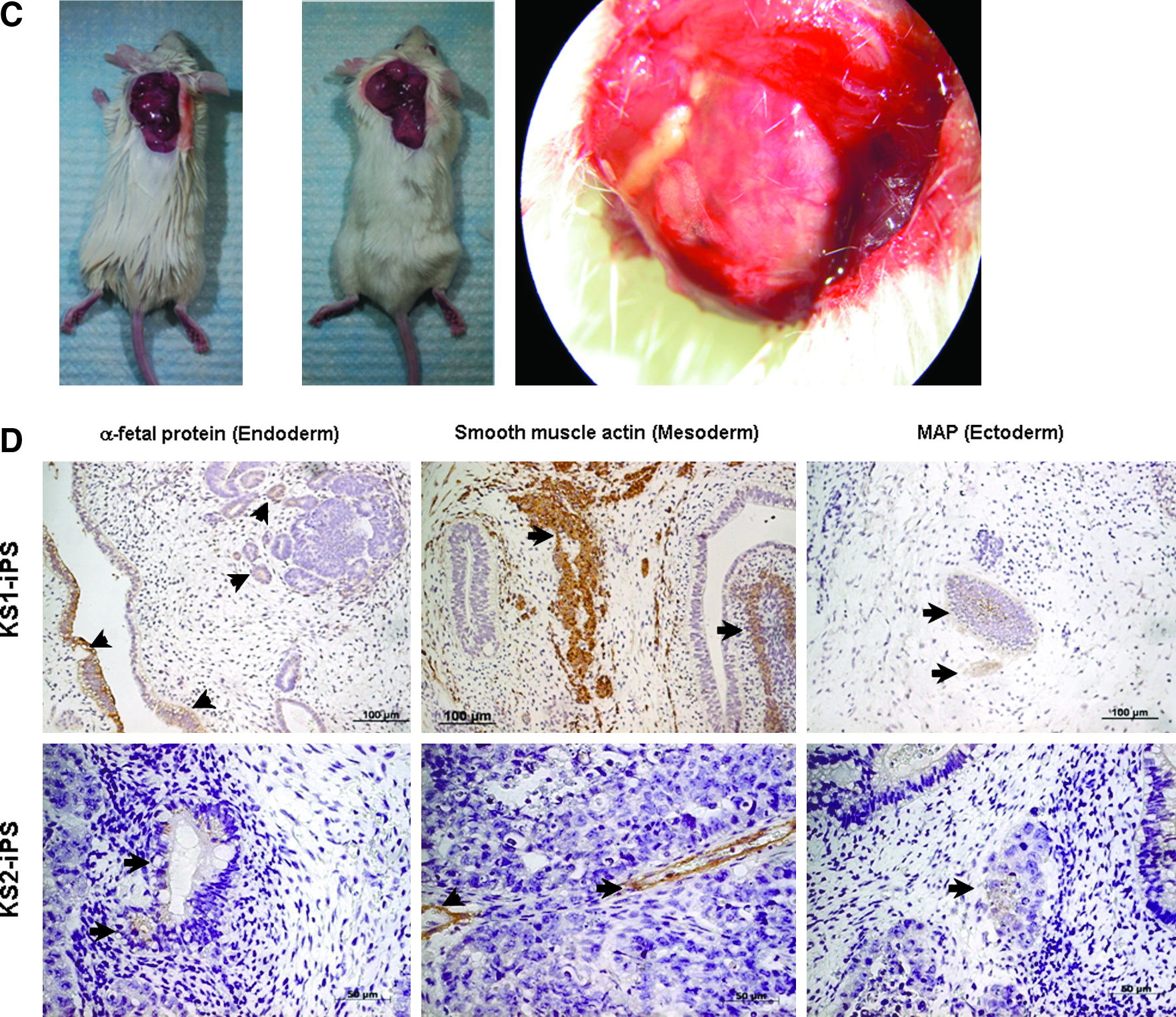
We next evaluate the pluripotency of these putative human iPSCs generated under feeder-independent, serum-free conditions. Upon differentiation induction by embryoid body formation with the hanging drop method, these putative human iPSCs were able to form embryoid bodies as embryonic stem cells, which could then be differentiated into the three primary embryonic germ layers after plating to gelatin-coated wells (mesoderm: smooth muscle actin; ectoderm: nestin; and endoderm: alpha-fetoprotein) (Fig. 6A). In addition, cardiomyocytes (troponin-I-positive cells) and endothelial cells [von Willebrand factor (vWF)-positive cells] were also observed within these differentiating embryoid bodies. Consistently, RT-PCR showed the expression of Indian hedgehog homolog (IHH), alpha-fetoprotein, intercellular adhesion molecule 1 (ICAM-1) and nestin with a reduced expression level of endogeneous pluripotency marker, REX1, in these differentiating putative human iPSCs (Fig. 6B). Furthermore, as teratoma-like mass formation after human embryonic stem cell injection to immunodeficient mice has been a standard assay to demonstrate developmental pluripotency, we transplanted these putative human iPSCs subcutaneously into six NOD/SCID mice. Six to 8 weeks posttransplantation, well-encapsulated tumors harboring differentiated tissues of the three embryonic germ layers were observed in five out of the six mice (Fig. 6C and D).
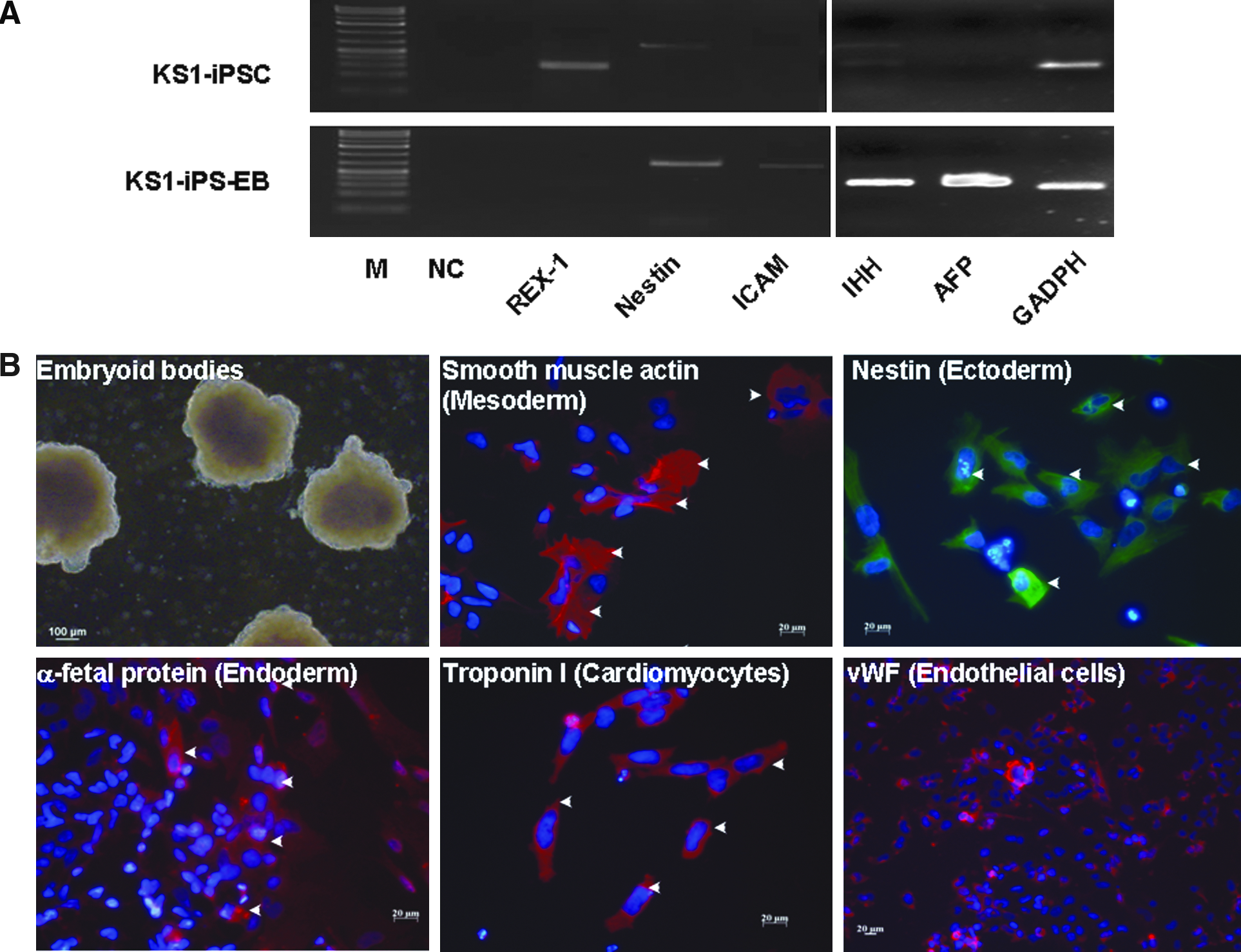
Differentiation of human iPSCs generated under feeder-independent, serum-free condition with defined medium.
In a separate experiment, these putative human iPSCs were induced to differentiate into EPCs with typical cobblestone morphology, ability of DiI-Ac-LDL dye uptake, and Ulex europaeus lectin antigen expression with standard protocol (Fig. 7A–F) (Vasa et al., 2001). In addition, these EPCs derived from the putative human iPSC line exhibited features including tube formation and migration typical of H1 hESC-derived EPCs (Fig. 7G).
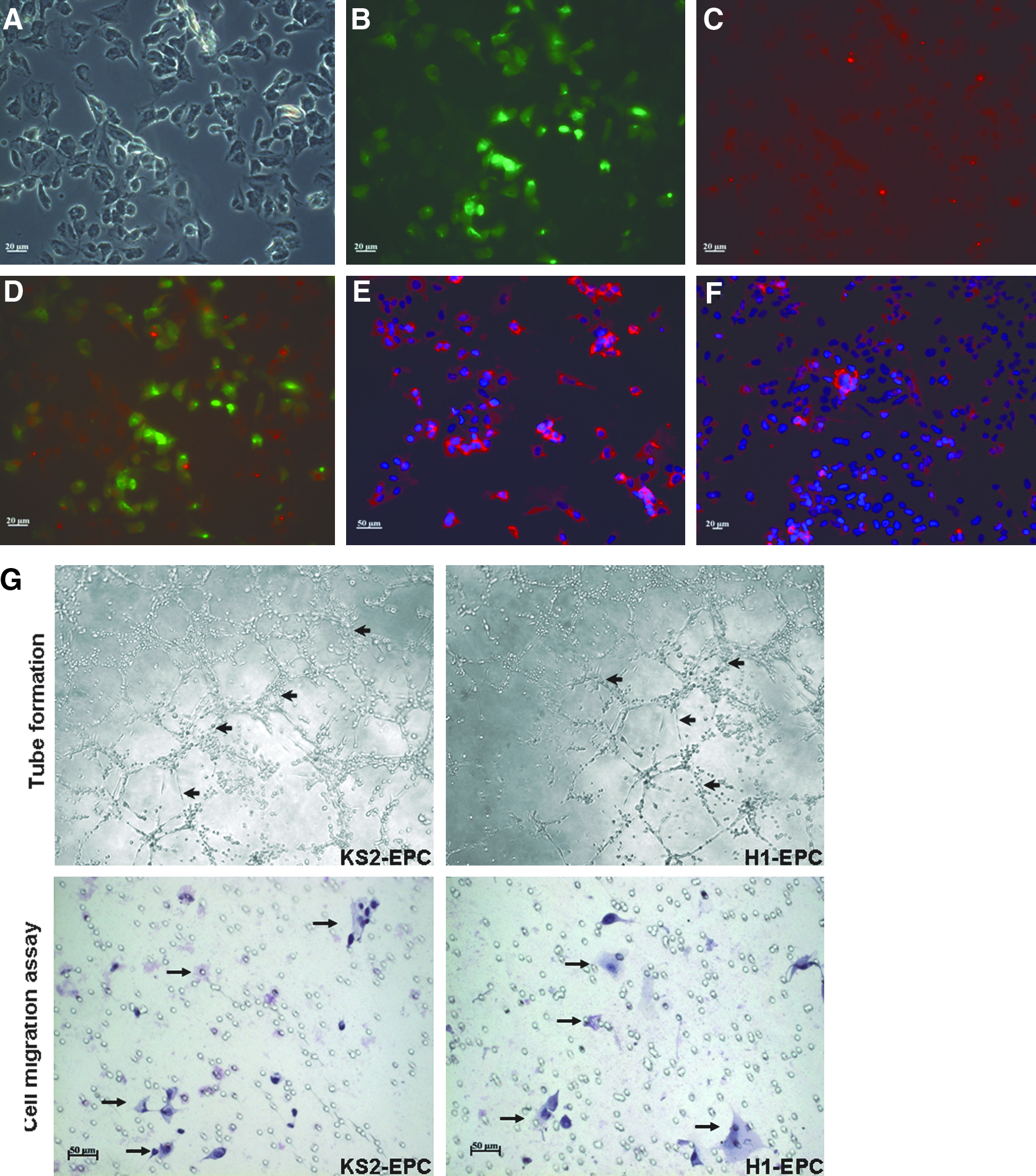
Differentiation of human iPSCs to endothelial progenitor cells (EPCs). Fluorescence staining analysis to characterize the KS1-iPSC-derived EPCs.
Discussion
In this study, we showed that human iPSCs can be readily generated from adult human dermal fibroblasts using lentiviral vector-mediated gene transfer of a set of transcription factors under feeder-independent, serum-free condition, which minimizes the risk of transmitting pathogenic agents of animal origin to human and reduces variability of reprogramming process related to the batch-to-batch variation of feeder cells. The established human iPSC lines retain normal karyotypes, and exhibit many features similar to human ESCs including morphology, pluripotency markers expression, hypomethylation of Oct-4 promoter regions, in vitro differentiation, and in vivo teratoma formation. The overall reprogramming efficiency as determined by the number of colonies per cells is approximately 0.01% to 0.02%, comparable to the highest efficiency of reprogramming adult human fibroblasts with the Yamanaka four factors reported in the literature (≈ 0.01%). Two salient features in our protocol have important implications in human iPSC generation including the use of ROCK inhibitor and the completely defined culture medium.
In our initial trial, we failed to generate any human iPSCs in the absence of a ROCK inhibitor despite documented successful viral transduction. Although ESC-like cluster formation was observed as early as 14 days posttransduction, these ESC-like clusters failed to expand and eventfully apoptosed. By contrast, the introduction of ROCK inhibitor at day 7 for 24 h, two human iPSC lines were successfully established. This suggests that ROCK inhibition plays a critical role in the generation of human iPSCs in the Matrigel and mTeSR system. One plausible explanation for the failure to generate human iPSCs with a feeder-independent system may be related to the fact that human iPSCs and ESCs, in contrast to their mouse counterparts, exhibit dissociation-induced apoptosis (Amit et al., 2004; Thomson et al., 1998). In the other words, human iPSCs and ESCs cannot survive and/or proliferate in single cells without feeder support. Thus, isolated human dermal fibroblasts after viral transduction may acquire this fragility during the reprogramming process, rendering them unable to proliferate in the absence of feeder. Recently, the selective ROCK inhibitor, Y-27632, has been reported to improve dramatically the survival of dissociated human iPSCs and ESCs in serum-free suspension without affecting their pluripotency (Baharvand et al., 2010; Braam et al., 2010; Li et al., 2009b; Nemati et al., 2010; Pakzad et al., 2010; Mollamohammadi et al., 2000; Watanabe et al., 2007). Although the mechanism remains incompletely understood, it is generally accepted that ROCK inhibitor exerts direct antiapoptotic effect on single human ESCs and iPSCs, thus attenuating the dissociation-induced apoptosis (Watanabe et al., 2007). In fact, there are emerging experimental evidences to suggest alternative mechanisms, from which ROCK inhibition supports undifferentiated growth of human ESCs and/or iPSCs in feeder-independent system (Harb et al., 2008; Krawetz et al., 2009; Pakzad et al., 2010; Peerani et al., 2007). For instance, in a series of publications from the Baharvand laboratory, ROCK inhibition has been shown to improve plating efficiency of human ESCs as well as iPSCs in a Matrigel-based, feeder-free culture system (Krawetz et al., 2009; Pakzad et al., 2010). More importantly, the expressions of a panel of integrins were upregulated in both human ESCs and iPSCs with ROCK inhibition, potentially medicating extracellular matrix adhesion and signaling, and extracellular matrix–cell interaction. Furthermore, ROCK inhibition has been shown to increase the OCT4 expression in human ESCs (Peerani et al., 2007). On the other hand, it has also been hypothesized that ROCK inhibition does not block apoptotic pathways in pluripotent stem cells; instead, it may render human ESCs less sensitive to the environmental changes (dissociation and detachment), thus allowing time for cell–cell interaction among these single cells to form aggregates (Krawetz et al., 2009). In fact, coadministration of ROCK inhibitor and EGTA, which disrupts E-cadherin-dependent cell–cell interaction, to dissociated human ESCs, abolishes the positive effects of ROCK inhibition (Li et al., 2009b). Taken together, we postulate that, although certain proportion of adult dermal fibroblasts after viral transduction were successfully reprogrammed to human iPSCs, they cannot survive and/or proliferate as single cells in the absence of cell–cell and cell–extracellular interaction. ROCK inhibition enhances the survival of these reprogrammed cells, thus improving the reprogramming efficiency. However, further experiments are needed to evaluate the relative importance of various pathways.
In addition, the use of a completely defined culture medium, mTeSR™1 medium with high levels of bFGF and TGF-β pioneered by Ludwig and coworker (2006), which removes the inherent batch-to-batch variability of feeders and conditioned media, allows more consistent condition for human iPSC generation, and improves its reproducibility. However, although the mTeSR™1 medium was completely defined, our system exploited here was not completely xenogeny free, which consisted of bovine serum albumin, matrigel, and trypsin, and the process of lentiviral production did involve other animal products. In fact, human iPSCs could now be generated with a reduced number of transgenes (Giorgetti et al., 2009; Li et al., 2009a; Nakagawa et al., 2008), adenoviral vectors (Zhou and Freed, 2009), nonintegrating episomal vectors (Yu et al., 2009), or even with reprogramming proteins (Kim et al., 2009) to prevent malignant transformation of human iPSCs. Further studies are required to combine of the feeder-independent, serum-free culture system with one of these nonlenti/retro-viral-based systems for generation of clinical grade patient-specific iPSCs. Nonetheless, our work is an important step toward generating clinical compatible patient-specific iPSCs to ultimate autologous iPSC-based therapy.
Footnotes
Acknowledgments
This work was supported by the Hong Kong Research Grant Council (HKU8CRF09 to Prof. Tse and Dr. Siu, and HKU 780110M to Prof. Tse).
Author Disclosure Statement
The authors have nothing to disclose.