
Abstract
Abstract
Human embryonic stem cells can be differentiated into insulin-secreting cells by emulating in vitro the key processes that occur during embryonic development. However, the resulting cells are generally immature; thus, further research must be performed to identify the necessary factors to complete the differentiation. To this end, we cultured mouse embryonic stem cells with pancreatic bud-conditioned medium, based on a recent publication. Unlike in humans, mouse cells present two types of insulin, which can be used to identify different cell lineages. As a result, the cell product presented a neuroectodermal genetic expression pattern, with no expression of any definitive endodermal marker analyzed. Also, nonglucose-dependent insulin release was detected. Altogether, this previously published protocol results in neuroectoderm, and not definitive endoderm, derived insulin-positive cells. This further confirms the difficulty of obtaining true cell types of this germ layer. Finally, we identified a 16-kDa protein band that was present in pancreatic bud-conditioned medium. Sequencing this band revealed the presence of Reg proteins. The role of pancreatic bud-conditioned medium remains to be tested in definitive endoderm committed cells.
Introduction
Altogether, these observations indicate that the current protocols must be further studied and possibly require additional factors and specific culture conditions to achieve an adequate in vitro differentiation. For example, several factors that appear during embryonic pancreatic development may also work in vitro, such as members of the fibroblast growth factor (FGF) family, activin and retinoic acid (D'Amour et al., 2006; J. Jiang et al., 2007; W. Jiang et al., 2007; Kroon et al., 2008; Nakanishi et al., 2007). These factors are released by several structures surrounding the future pancreas, such as the mesodermal plaque, notochorda and aorta, as well as from the pancreatic developing compartment (Murtaugh and Melton, 2003). In this context, several protocols use coculture and cotransplantation strategies in order to mimic pancreatic differentiation (Banerjee et al., 2010; Brolén et al., 2005; Chen et al., 2009). Specifically, a recent publication claimed to have produced definitive endoderm-derived, insulin-secreting cells by culturing mouse ESCs with embryonic pancreatic rudiment-conditioned medium (Soria et al., 2008; Vaca et al., 2006). The aim of the present study was to investigate in detail the origin of the insulin-containing cells that can be obtained with this approach. Also, because this protocol uses pancreatic rudiment-conditioned medium, we isolated and identified several soluble factors present in this medium.
The major discrepancies found between this study and the published protocol (Soria et al., 2008; Vaca et al., 2006 2008) are the following. In the differentiation phase, it is impossible to obtain isolated and spheroid cell aggregates, known as embryoid bodies (EBs), with the concentration of cells suggested in the publication (Vaca et al., 2006, 2008). Also, in the cell selection phase, the authors do not explain how they obtain transfected cells that are capable of resisting such a high antibiotic concentration (Vaca et al., 2006, 2008). Our personal experience showed that at this concentration all the cells in culture entered apoptosis, and conversely to Vaca et al. (2006), we were not capable of detecting cell proliferation after antibiotic selection. Furthermore, we have proven with solid evidence (RT-PCR, immunocytochemistry, and insulin secretion), that the obtained cells derive from the neuroectoderm and primitive endoderm lineages. In no case did we obtain cells from the definitive endoderm lineage as stated by the authors (Soria et al., 2008). The expression of Pdx-1 seems to be unnecessary in our protocol. In fact, we have obtained the same differentiation results by using a batch of D3-ESCs Pdx-1+ (Vaca et al., 2006), and with another batch of D3 cells Pdx-1−. Finally, in this study insulin release seems to be independent of extracellular glucose concentration, contrary to the hormone release published by Vaca et al. (2006).
Materials and Methods
Cell culture, generation of pancreatic bud-conditioned medium, and in vitro differentiation protocols
Mouse D3-ESCs were purchased from ATCC (American Type Culture Collection, Barcelona, Spain). Cells were cultured in D3 culture medium (D3-CM): high glucose DMEM (Dulbecco's Modified Eagle's medium) (Gibco, Grand Island, NY) as described in Vaca et al. (2006). Undifferentiated cells (8 × 106 cells) were transfected by electroporation (Electro Square Porator ECM830, CA) at 250 volts for 1 msec with 24 μg of a similar KpnI linearized DNA construction used by Vaca et al. (2006). Transfected cells (33% efficiency) were selected by adding 200 μg/mL hygromycin (Calbiochem, La Jolla, CA) and maintained in the same culture conditions in the presence of leukaemia inhibitory factor (LIF) (Chemicon, Temecula, CA).
Four differentiation protocols were performed using control conditions (A1/B1) or in the presence of a conditioned medium (A2/B2). In both cases, the cells were cultured at high density (A1/A2), as in Vaca et al. (2006), and also using a lower cell density (B1/B2) (see Table 1).
D3-CM, D3-culture medium; PBCM, pancreatic bud conditioned medium.
For the generation of pancreatic bud-conditioned medium, we used the procedure described in Vaca et al (2006). The animals were manipulated according to the regulations and under supervision of the Ethics Committee of Miguel Hernandez University. The resulting medium, which contained the factors released by the pancreatic buds, was diluted 1:1 with D3-CM and stored at −80°C until use. This medium was called pancreatic bud conditioned medium (PBCM).
To induce differentiation (protocols A1 and A2), 1.5 × 106 cells/mL were cultured for 7 days in nonadherent bacterial plates with either D3-CM (A1) or PBCM (A2), supplemented with 3% fetal calf serum (FCS) with no LIF. As a result, the floating cells formed large clusters, which were placed in adherent plates for another 7 days in the same medium but adding 10% FCS instead of 3%. Afterward, the cells were cultured with 2.3 mg/mL G-418 (Gibco) overnight to select the insulin-expressing cells. The following day, the cells were washed extensively to eliminate the dead cells. In protocols B1 and B2, differentiation was induced by performing low density cell cultures (5 × 104 cells/ml) to form EBs in 3% FCS with no LIF, in the presence of D3-CM (Protocol B1) or PBCM (Protocol B2) for 7 days. Cells were then transferred to adherent plates for an additional 7 days in the same medium with 10% FCS. Finally, insulin-producing cells were selected by incubating overnight with 0.2 mg/mL G418.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
RNA was extracted from undifferentiated and differentiated cells using the Tripure reagent (Roche, Basel, Switzerland) according to manufacturer's instructions. Total RNA (1 μg) was reverse transcribed using reverse transcriptase (Roche) and amplified with a Taq polymerase kit (Roche). Reactions containing specific primers were carried out in a final volume of 25 μL at specific annealing temperatures as indicated in Table 2. Four independent experiments were performed in duplicate, using β-actin as control.
Immunocytochemistry
Cells were washed with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde at 4°C for 16 h, then washed and permeabilized with 0.05% Triton X-100 (Sigma, Steinheim, Germany) at 4°C for 6 h. At the end of the process, cells were washed twice with PBS and blocked with 10% goat serum and 3% bovine serum albumin (BSA) (Sigma) at 4°C for 1 h for simple immunostaining, and 10% donkey plus 10% rabbit sera for double immunostaining. Afterward, the cells were incubated overnight with one of the following primary antibodies: goat anti-C-peptide (1:100, Linco, St. Charles, MO) and chicken antineurofilament middle (1:500, Stemcell, Vancouver, BC). The next day, several washings were performed with PBS before incubating with the secondary antibody. The secondary antibodies used were: antigoat IgG fluorescein isothiocyanate (FITC) (1:500, Jackson, West Grove, PA) when performing simple immunostaining, or antigoat IgG Cy3 (1:1500, Jackson) in double immunostaining, and antichicken IgY FITC (1:500, Abcam, Cambridge, UK). Control cells incubated with only the secondary antibodies showed no staining. Finally, cell nuclei were counterstained with 1 mg/mL Hoescht 33258 (Sigma) for 3 min in darkness at room temperature. Coverslips were washed for 5 min with PBS in darkness and fluorescence was preserved at −80°C with the Vectashield kit (Vector, Burlingame, CA). Samples were observed by fluorescence microscopy (Olympus AX70, PA) and analyzed by DP-SOFT 3.2 software (Olympus).
Insulin content and secretion
Cells (2.5 × 105/well) were incubated in 24 multiwell plates (TPP, Transadingen, Switzerland) in the presence of D3-CM supplemented with either 10% FCS or PBCM. Cells were washed three times and incubated for 24 h with continuously gassed Krebs buffer pH 7.4 containing 0.1% BSA and 5 mM glucose. Incubation buffer was discarded and cells incubated again in the presence of 3 or 22 mM glucose for an additional hour. At the end of this period, the buffer was collected and centrifuged for 5 min at 800 × g. Approximately 80% of the supernatant was frozen at −80°C. To analyze insulin content, the attached cells were resuspended in 500 μL of 72% ethanol: 0.2 N HCl for 16 h at 4°C and frozen afterward at −80°C. Insulin was determined by radioimmunoassay (RIA) using the Coat-A-Count kit (Siemens, CA). INS-1E (passage ∼60) cells were used as positive controls. Secretion was normalized to total insulin content. Data was represented as mean ± standard error of the mean (SEM). Student's t-test was conducted to determine statistical significance.
SDS-Polyacrylamide gel electrophoresis and protein band sequencing
D3-CM and PBCM (40 μg protein/lane) were placed in SDS-polyacrilamide gels in 8% acrylamide, according to standard procedures. A characteristic band of 16 kDa was repeatedly detected after Sypro Ruby (BioRad, Hercules, CA) staining only in the PBCM lane. The band was cut and digested with trypsin before mass spectrometry analysis.
Results
Due to previous discrepancies in reproducing several published protocols to generate insulin-secreting cells, we decided to study one of the most recent approaches using the differentiation potential of soluble factors released by the pancreatic rudiments (Vaca et al., 2006) in mouse ESC in which the different types of insulin can be further analysed (Protocol A2, Table 1). In the first step of differentiation, where EBs were formed, there was a remarked morphological difference depending on the initial cellular density (Table 1). Specifically, at high cell density the EBs were large and irregular shaped, compared to the small spheroid structures detected at lower densities (Fig. 1). To reproduce the published protocol (Vaca et al., 2006), it was necessary to perform several transfections (5–10) on the same population of cells to obtain the antibiotic resistance mentioned (Vaca et al., 2006). In parallel, we used one transfection round and selected cells with 10 times less antibiotic than the referred report by Vaca et al. (protocols B1 and B2). It must be mentioned that the addition of G418 provoked massive cell death and that its presence inhibited cell proliferation in all the protocols assayed. Finally, in order to test the role of PBCM (protocols A2 and B2) in the differentiation to insulin-secreting cells, we cultured D3 cells with D3-CM (protocols A1 and B1) and the resulting cells were characterized.
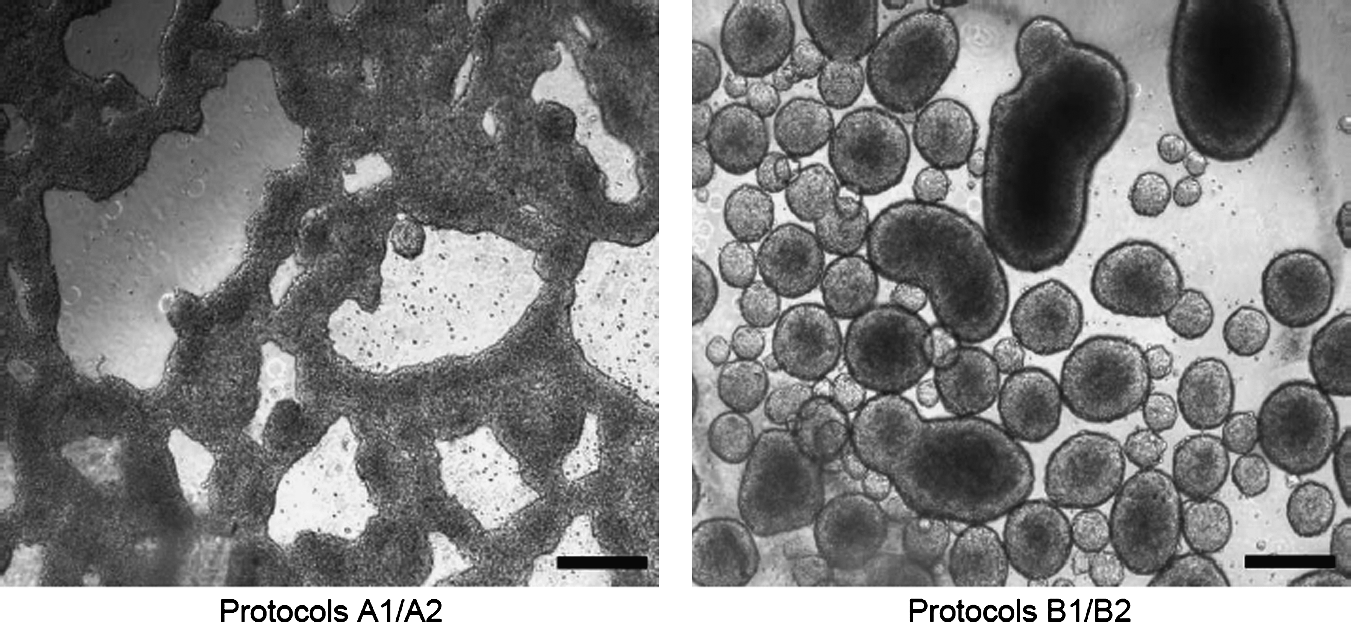
Phase contrast images showing cell aggregates obtained in nonadherent plates using 1.5 × 106 cells/mL (Protocols A1 and A2) or 5 × 104 cells/mL (Protocols B1 and B2). Bar: 100 μm.
Total RNA was extracted and RT-PCR analysis performed (Fig. 2). Oct4 gene expression was detected in all the assayed protocols, indicating the presence of an undifferentiated precursor population and thus confirming previous results by our group (Enseñat-Waser et al., 2006). The presence of Amnionless (Amn) and Neurofilament middle (NefM), specific primitive endoderm and neuroectoderm markers, respectively, suggested that in all protocols there was a significant population of cells from these lineages. Also, PBCM seemed to increase the expression of these markers in A2 and B2. The expression of insulin II gene, which is also expressed in these lineages, confirms these observations. The expression of insulin I and II would indicate that the cells differentiate toward definitive endoderm; however, insulin I was not detected in any of the protocols. Other genetic markers (not shown), such as Brachyury (Bry), a mesendoderm marker, and Cxcr4, a marker for definitive and primitive endoderm, were not detected when cells were incubated in the presence of PBCM. Only a faint glucagon signal was detected under these culture conditions in the presence of PBCM (not shown).
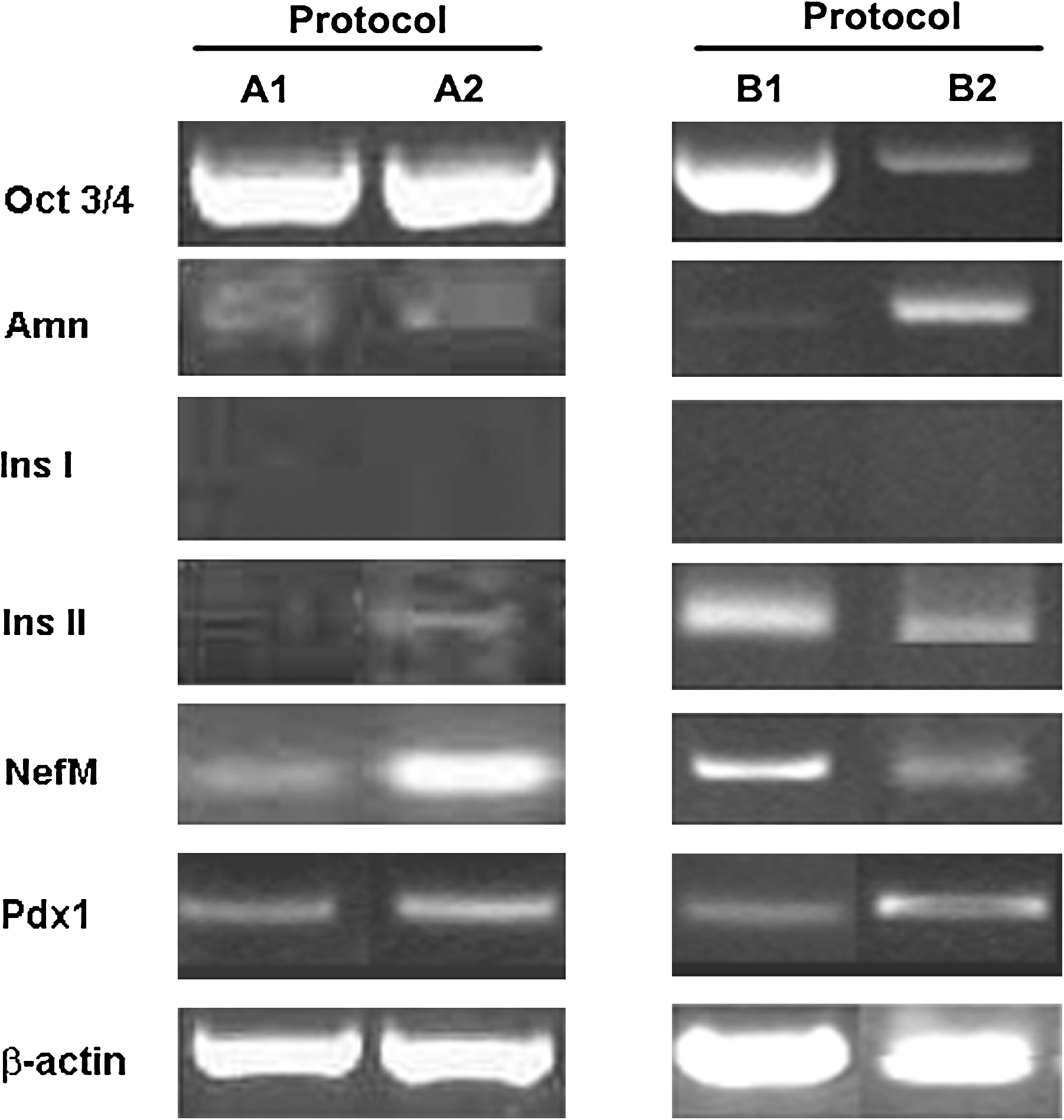
RT-PCR analysis of neomycin-selected cells according to protocols A1/A2 and B1/B2. Undifferentiated hygromycin-resistant D3 cells were induced to differentiate by forming cell aggregates (1.5 × 106 cells/ml for protocols A1 and A2, or 5 × 104 cells/mL for protocols B1 and B2), plated in monolayer (outgrowth phase) and G418 selected (2.3 mg/mL for protocols A1 and A2, or 0.2 mg/mL for protocols B1 and B2). Total RNA was extracted from cells and analyzed by RT-PCR. β-Actin was used as control.
Altogether, our results indicate that these protocols give rise to insulin-positive cells of neuroectoderm and primitive endoderm lineages, but not from definitive endoderm, the cell lineage from which pancreatic β-cells derive. The results obtained by RT-PCR analysis in protocols A1 and A2 were validated by immunocytochemistry experiments. The majority of the cells cultured with D3-CM or PBCM coexpressed NefM and C-peptide (Fig. 3), with very few cells expressing only C-peptide. This double staining indicated that the cells derived mainly from neuroectoderm and in a minor proportion from the primitive endoderm lineage. However, this could not be further studied as there was no commercially available Amn antibody. The cells from protocols B1 and B2 presented similar expression patterns as A1 and A2, respectively. In all protocols, the presence of C-peptide indicated that insulin was synthesized de novo by the differentiated cells, thus discarding that insulin could be taken from the culture medium (Rajagopal et al., 2003).
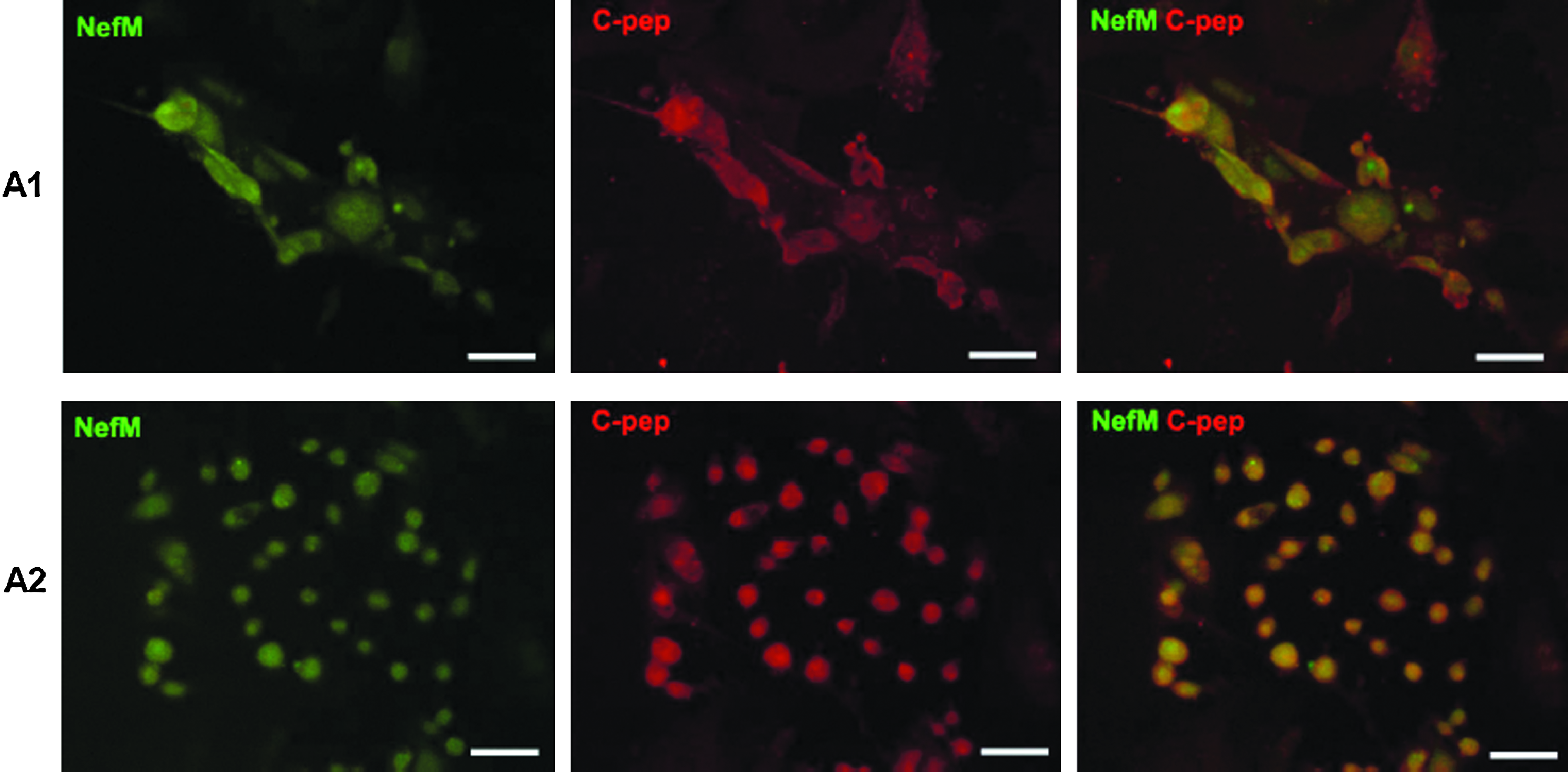
Neomycin-selected cells from protocols A1 and A2 coexpress neuroectodermal markers. Fluorescence images of NefM (green) and C-peptide (red) cells (merge: NefM/C-pep). Scale bar = 100 μm.
In order to test the functionality of the resulting cells, insulin secretion experiments were performed. After antibiotic selection, cells were washed carefully to eliminate dead cells and debris. As it can be seen in Figure 4, the cells obtained in the various protocols released low amounts of insulin and did not respond to high glucose concentrations. As a positive control we used INS-1E cells cultured in the presence of D3-CM or PBCM, which resulted in two to three times more insulin secreted in the presence of high glucose (Fig. 4).
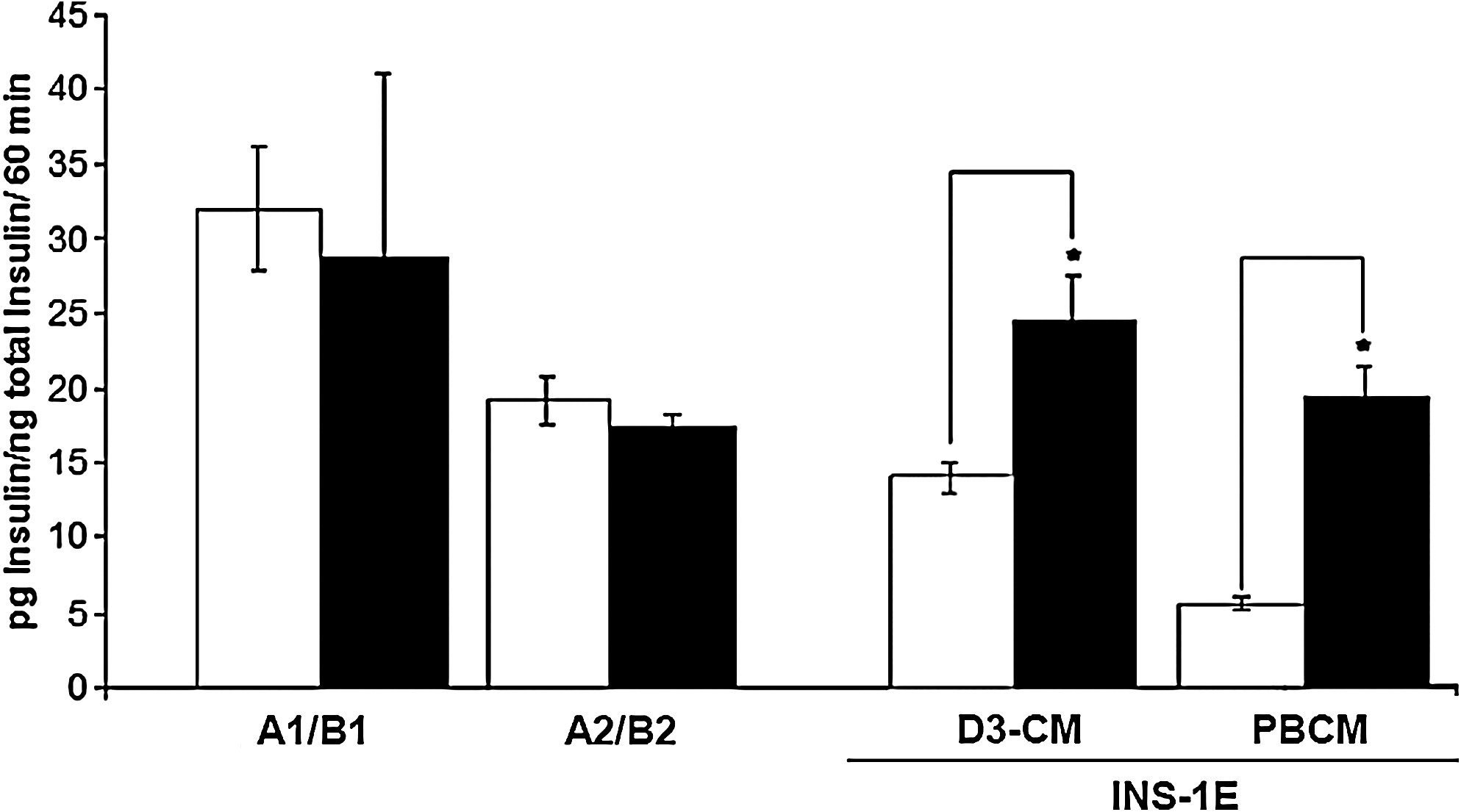
Insulin secretion of G-418-selected cells (Protocols A1 and A2) and INS-1E cells in response to 3 mM glucose (open bars) and 22 mM glucose (black bars) under different culture conditions: D3-CM and PBCM. Each value represents mean ± SEM of four independent experiments. Statistical significance was tested by Student's t-test, *p < 0.05.
In order to discard a cell batch-specific aberration, we used different batches of D3 cells from ATCC (Rockville, MD), obtaining the same previously shown results. In one of these batches, we could reproduce all the differentiation process but with no Pdx-1 expression, suggesting that the expression of this transcription factor does not seem to be necessary for the differentiation towards insulin-producing cells. Interestingly, same observation was noticed in one protocol that derives induced-pluripotent stem (iPS) toward pancreatic β-like cells (Alipio et al., 2010).
One of the main objectives of the present work was to identify the factors in PBCM that could favour the differentiation of D3-ESCs, in this particular case toward neuroectodermal and primitive endoderm insulin-positive cells. To this end, we performed a simple characterization of the PBCM versus D3-CM by SDS-PAGE. As a result, we observed a prominent and repetitive protein band at the region of 16 kDa in the conditioned medium that was not present in D3-CM (Fig. 5). Purification and subsequent sequencing revealed three candidate peptides corresponding to chymotrypsinogen B1, secreting granule protein zymogen 16 and Reg1. The two first peptides are expressed in the exocrine pancreas, whereas Reg1 is a protein implicated in tissue regenerative processes in neuroectoderm derived tissues (Nishimune et al., 2000) as well as in the endocrine pancreas (Terazono et al., 1990).

Proteins (40 μg) from D3-CM and PBCM were analyzed by SDS-PAGE (8% acrylamide). Figure shows a typical silver stained gel. The arrow indicates a band that is present at the 16-kDa region in the PBCM but not in D3-CM. MWM: Molecular weight markers.
Discussion
A major concern in protocols to differentiate ESCs toward insulin-producing cells is to recognize the true origin of these cells, as insulin is expressed in tissues from different lineages such as the neuroectoderm, primitive endoderm, or definitive endoderm. To this end, recent protocols have strived to obtain definitive endoderm-derived cells (D'Amour et al., 2005; Yasunaga et al., 2005), which can be further differentiated into pancreatic insulin-secreting cells (D'Amour et al., 2006; Kroon et al., 2008). As we have shown here, a previously published protocol resulted in obtaining nonpancreatic cells (Vaca et al., 2006).
In this context, there are two points to mention in this published procedure that deserve some attention to reproduce this protocol in other laboratories. The first refers to the number of cells initially used to form EBs. Other protocols generally use a lower cellular density, between 103 and 105 cells/mL (Enseñat-Waser et al., 2006; Ku et al., 2004; McKiernan et al., 2007; Moritoh et al., 2003; Nakanishi et al., 2007; Shim et al., 2007), whereas in the referred protocol the cells were cultured at a density of 1.5 × 106 cells/mL (Vaca et al., 2006). As a result, instead of the floating independent spheroid structures that are usually observed in these types of cultures (Enseñat-Waser et al., 2006), the cell clusters were very large and irregular shaped (Fig. 1). We were not able to find any previous study concerning this type of structures, nor if they constitute an appropriate in vitro differentiation condition.
In addition, for certain commercialized cell lines, such as D3 and R1-mouse ESCs, the expression of definitive endoderm markers occurs very late in EB cultures. For example, we have observed spontaneous glucagon gene expression in EB cultures at day 14 (Roche et al., 2007) and Cxcr4 at day 30 (not shown). The addition of PBCM could possibly accelerate the differentiation processes in cell aggregates (protocol A2) or EBs (protocol B2), However, our results indicate that the factors present in this conditioned medium induce a very modest differentiation.
A second point to mention concerns the amount of G-418 used to select insulin-positive cells in the referred publication (Vaca et al., 2006). We have also noticed in the protocol the use of an unusually high dose of G418 antibiotic for cell selection. In our observations, this high concentration of G418 resulted in total cell death. In previous reports, 200 μg/mL neomycin was used to select the cells containing the same plasmid (Soria et al., 2000). However, Vaca et al. used over 10 times more antibiotic (2.3 mg/mL). Because this protocol is used again in a more recent publication of the same group (Vaca et al., 2008), this would suggest the use of a particular cell clone capable of resisting the large amount of antibiotic. In an attempt to reproduce these results, we performed consecutive transfections in order to obtain cells with the same antibiotic resistance. Throughout our attempts, we observed that the selected cells, independently of the antibiotic dose, presented a similar genetic expression pattern. Thus, cell selection with higher concentrations of G418 does not result in a more differentiated cell than using a normal dose of the antibiotic (0.2 mg/mL compared to 2.3 mg/mL).
Furthermore, in our hands the antibiotic-based selection resulted in obtaining a population of cells that did not continue proliferating. Unlike the data shown in the previous publication (Vaca et al., 2006), we did not observe BrdU incorporation in the selected cells when G418 was present in the culture medium. On the contrary, we observed a high number of apoptotic cells, detected by Hoechst staining, directly proportional to the antibiotic dose used. This is a very important point to be considered when performing insulin secretion experiments, as the dead cells can release insulin in an unspecific manner. This could explain the difference in secretion detected in the present study compared to that observed in Vaca et al. (2006). Additionally, the present work detected a lack of glucose-induced insulin secretion, which could initially be explained by the presence of inhibitory factors in the PBCM. However, our results with INS-1E indicate otherwise, because insulin secretion was similar in these cells when incubated with D3-CM or PBCM, supporting the presence of a nonregulated release of insulin in the mouse D3-ESC obtained through these protocols. Additionally, we detected the expression of NefM and Amn, suggesting neuroectoderm and primitive endoderm lineages, respectively. Insulin II, which was also detected, is expressed in both these embryonic layers as well (Milne et al., 2005; Roche et al., 2005). However, we did not detect insulin I, Bry, or Cxcr4, which are definitive endoderm markers. The presence of preproglucagon in PBCM cultures could suggest the presence of some definitive endoderm-derived cells. Preproglucagon is expressed in definitive endoderm-derived intestinal cells as well as in neuroectoderm-derived tissues, giving rise in both cases to glucagon-like peptide I (GLP-1). We thoroughly characterized these glucagon-positive cells and observed that under this differentiation protocol these cells derived from the neuroectoderm (Roche and Vicente-Salar, manuscript in preparation). This observation is supported by the absence of the specific intestine marker Cadherin 17 (not shown).
C-peptide is the main marker for de novo insulin biosynthesis. However, this protein can not be used to indicate a pancreatic lineage, as neuroectodermal insulin-positive cells are also C-peptide positive (Sipione et al., 2004). Our data obtained from the immunocytochemistry assays confirm this point. The presented data suggest that PBCM contains soluble factors that could favour the differentiation of D3-ESCs to neuroectoderm. In this sense, we detected the presence of Reg-1 protein in PBCM. This factor forms part of a large family of peptides involved in regeneration and proliferation of several cell types (Nishimune et al., 2000; Simon et al., 2003; Terazono et al., 1990). For example, Reg-1 and Reg3δ are involved in β-cell regeneration and embryonic development (Hamblet et al., 2008; Fleming and Rosenberg, 2007). Reg3β seems to be involved in hepatic regeneration as well as have a neurotrophic effect (Nishimune et al., 2000; Simon et al., 2003). Reg1 is present during pancreatic development at e8–e12 (Perfetti et al., 1996), supporting the presence of Reg1 in the conditioned media obtained from pancreatic rudiment cultures.
Altogether, these results support a possible role of pancreatic bud culture-conditioned medium in promoting neuroectoderm differentiation in D3-ESCs, but not to definitive endoderm as it has been previously published (Soria et al., 2008; Vaca et al., 2006). Therefore, the potential of this coaxial strategy in a cell system committed to definitive endoderm remains to be tested.
Footnotes
Acknowledgments
This work was funded by grants to ER from Conselleria de Sanitat de la Generalitat Valenciana (AP-100/09), Fundacion Medica Mutua Madrileña, Instituto de Salud Carlos III-FEDER (PS09/01093), and Fundacion Salud 2000-Merck Serono.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.