
Abstract
Abstract
The cloning of animals by somatic cell nuclear transfer (SCNT) has the potential to allow rapid dissemination of desirable traits from elite animals. However, concern has been expressed that aberrant epigenetic marks in SCNT-derived animals may be passed onto the next generation, even though the offspring of clones appear to be mainly normal. Here, we compared the DNA methylation patterns at 10 genomic regions in sperm from SCNT bulls with that from normal, naturally conceived bulls and with the nuclear donor somatic cells. Eight of the 10 genomic regions were differentially methylated in sperm compared with the donor cell DNA. All three satellite sequences examined here were less methylated in sperm than in the donor cells, contradicting the belief that the sperm genome is always highly methylated. The DNA methylation patterns at all 10 regions were almost identical between SCNT and control sperm, with only one out of the 175 CpG sites/groups of sites examined showing significant difference. These results provide the first molecular evidence that the donor cell genome is correctly reprogrammed upon passage through the germ line in males, and that any epigenetic aberrations harbored by SCNT bulls are unlikely to be passed onto their offspring.
Introduction
Based on reports from a small number of studies (Alexander et al., 2007; Ortegon et al., 2007; Shimozawa et al., 2002; Wells et al., 2004), the progeny of clones do not show the poor postnatal health encountered with the original cloned parents, and they display normal growth parameters, blood profiles, reproductive capabilities, and normal telomere lengths. Thus, to date, it appears that the SCNT-associated phenotypes are not transmitted to the offspring following natural mating of cloned animals. These findings suggest that the developmental problems experienced with clones are epigenetic effects that appear to be corrected during gametogenesis. However, concerns remain that the progeny of cloned animals may still harbor some of the epigenetic defects from their clone parents after transmission from their gametes. Although epigenetic errors have been reported in SCNT-derived embryonic, fetal, and new-born tissues (Cezar et al., 2003; Couldrey and Lee 2010; Dindot et al., 2004; Long and Cai 2007; Lucifero et al., 2006; Sawai et al., 2009), to date, there are no reported studies that have examined the epigenetic status of mature gametes produced by clones.
DNA methylation is erased in male primordial germ cells and is reestablished during the process of spermatogenesis (Hajkova et al., 2002; Reik et al., 2001). Demethylation, de novo methylation, and accurate maintenance of methylation processes are therefore all required with every life cycle to ensure proper erasure, acquisition, and maintenance of DNA methylation marks. The effects of a demethylating agent, 5-azacytidine, in rats have provided evidence of a link between abnormal DNA methylation, altered fertility, and abnormal embryo development (Oakes et al., 2007). Even if methylation errors in sperm are not sufficient to reduce fertility, their presence remains a concern as recent studies suggest that DNA methylation abnormalities present in germ cells may persist in future generations (Kobayashi et al., 2009; Lee et al., 2009; Stouder and Paoloni-Giacobino 2009; Stouder et al., 2009). Studies are therefore required to determine whether any existing epigenetic reprogramming errors persist in the gametes produced by cloned animals. In this study, we examined DNA methylation patterns in 10 genomic regions in mature spermatozoa produced by bulls generated by SCNT and compared the patterns with that of normal bull sperm. DNA methylation patterns of nuclear donor somatic cells were also examined to assess the extent of reprogramming after SCNT and gamete formation.
Methods
Production of SCNT bulls
All manipulations of animals involved in the present study were conducted in accordance with the regulations of the New Zealand Animal Welfare Act of 1999. SCNT embryos were produced essentially as described (Lee et al., 2004). An adult skin fibroblast cell line (AESF-1) from a high genetic merit Friesian bull was used as nuclear donors. SCNT bull calves were reared with their surrogates until weaning and then grazed on pasture, as per normal farm practice in New Zealand. Semen collection was initiated at 15 months of age and a second collection was made 3 months later. Only samples from the second collection were used in the DNA methylation analysis.
Semen collection and DNA extraction
Semen was collected from SCNT (n = 3) and control (n = 4) bulls using an artificial vagina and teaser heifers, according to the commercial practice of Animal Breeding Services, Ltd. (Hamilton, New Zealand). Three of the control bulls were from a commercial livestock breeding company and were of proven fertility. The fourth was a contemporaneous control bull generated by artificial insemination at the same time as the SCNT bulls were generated and was run in the herd with them. Semen was also collected from a further 14 SCNT bulls derived from four additional somatic donor cell lines carrying different genetics.
The volume of the ejaculate was measured and sperm number, viability, and motility were assessed, as per commercial practice. Sperm from approximately 250 μL of semen were washed with 4 × 1 mL phosphate-buffered saline (PBS). The resulting pellet was resuspended in 0.2 mL PBS/2% SDS with 200 μg/mL proteinase K and incubated at 60°C for 4 h. Sperm were then pelleted and resuspended in 0.1 mL PBS/2% SDS and 0.1 mL lysis buffer (1 mg/mL proteinase K, 55 μM DTT, 80 μM EDTA) and incubated at 60°C overnight. DNA was then extracted with one volume of buffered phenol/chloroform and precipitated from the aqueous phase with two volumes of 100% ethanol and 0.1 volume of 3M sodium acetate. Precipitated DNA was washed twice in 70% ethanol, air dried and redissolved in 100 μL water.
Somatic cell DNA extraction
DNA was extracted from the donor somatic cells used for SCNT using the DNeasy kit, following the manufacturer's protocol (Qiagen, Austin, TX). The cells were cultured in DMEM/F12 with 10% fetal calf serum (FCS), as described previously (Oback and Wells, 2003). DNA was extracted from approximately 106 cells using standard proteinase K digestion followed by phenol/chloroform extraction and ethanol precipitation.
Restriction digestion with methylation-sensitive enzymes
Six micrograms of sperm genomic DNA from a control bull was digested overnight at 37°C in a 200-μL volume with 80 U of the methylation-sensitive restriction enzyme HpaII or its methylation insensitive isoschizomer MspI. The digest was purified by phenol/chloroform extraction and precipitated using 0.1 volume 3 M sodium acetate and 2.5 volumes of 100% ethanol. Samples were resuspended in 20 μL water and visualized by electrophoretic separation on a 1% TBE agarose gel and stained with ethidium bromide.
Analysis of DNA methylation
Primer design
Primers were designed using MethPrimer to flank and amplify CpG island sequences, after bisulfite treatment of DNA, as described (Couldrey and Lee, 2010; Li and Dahiya, 2002). The genomic regions of interest included: (1) a region spanning the putative transcription start site of ASCL2; (2) a region in exon 10 of IGF2 (GenBank accession no. X53553) that is differentially methylated (DMR) in bovine gametes (Gebert et al., 2006); (3) 500 bp of the CpG island corresponding to the human KCNQ1OT1; (4) 0.5–1 kb upstream of exon 1 of HAND1; (5) a region beginning upstream of exon 1, covering exon 1 and part of intron 1 of SNRPN (GenBank accession no. AY743660); (6) a CpG island at the transcriptional start site of DKK-1; (7) a region in the glucocorticoid receptor (GR) equivalent to that previously shown to be epigenetically modified by maternal behavior toward the offspring in rat pups (Szyf et al., 2005; Weaver et al., 2004, 2005); (8) a region spanning a CpG island in satellite I sequence (GenBank accession no. J00032); (9) a region spanning a CpG island in satellite II sequence (GenBank accession no. X03116); and (10) a region spanning a CpG island in satellite alpha sequence (GenBank accession no. AJ293510).
MassArray analysis
DNA samples were analyzed using the methods described (Coolen et al., 2007; Couldrey and Lee, 2010; Ehrich et al., 2005). Briefly, 1 μg DNA was bisulfite treated using the EZ-96 DNA Methylation gold kit (Zymo, Orange, CA, USA) to produce methylation-dependent sequence variations of C to T and regions of interest were amplified using T7 tagged PCR primers. PCR products were analyzed by agarose gel electrophoresis to confirm successful amplification. In vitro amplification and transcription was performed on the reverse strand by T7 DNA and RNA polymerases and a simultaneous U specific cleavage by RNAse A. Approximately 20 nL of each sample was spotted onto Sequenom MassArray chips and subject to mass spectrometry. The efficiency of bisulfite conversion was determined by assessing the quality of the raw data. This protocol enables precise and accurate high-resolution, high-throughput DNA methylation analysis, quantitative to 5% methylation for informative CpG dinucleotides (Coolen et al., 2007).
Statistical analysis
The relative methylation of each of the CpG sites/group of sites was calculated (EpiTYPER, Sequenom, San Diego, CA, USA) by dividing the peak intensity (area under the peak) of the fragment representing the original methylated DNA, by the sum of the intensities of the peaks representing both methylated and nonmethylated DNA. Bartlett's test for homogeneity was used to compare standard deviations at different CpG sites within each genomic region. Due to the high significance of Bartlett's test, CpG sites were analyzed independently after angular transformation using a one-way ANOVA followed by individual comparisons, using pooled standard deviations for each CpG site. Storey-Tibshirani adjusted q was used to estimate true nulls (false discovery rate) (Storey and Tibshirani, 2003). Results from ANOVA and false discovery rate analysis were compared with results from the Bonferoni test to ensure significance. Results are presented as mean ± standard error of the mean (SEM).
Results
Semen samples were assessed by the commercial company for volume, sperm numbers, motility, and percentage of live versus dead sperm. The mean volume of the ejaculate for the SCNT bulls was 2.6 mL, similar to the volume of normal bull semen, which can vary between 1.5 and 5 mL. The percentage live sperm varied between 60 and 90% with motility scores that were not different between SCNT and control bulls. Sperm numbers per milliliter ejaculate were similar between two of the SCNT bulls and control animals. In the third SCNT bull, the sperm number was lower on both collections.
The DNA methylation patterns for each of the genomic regions in the nuclear donor cells and from sperm are shown in Figure 1. For each of the regions, not all CpG sites or groups of sites could be analyzed by this method; the number of CpG sites able to be analyzed are listed in Table 1. Where a fragment contains two or more CpG sites, the methylation level of that group of sites is that of the most methylated site in the group. Where two fragments cannot be distinguished from each other (Table 1), the methylation levels are averaged across the fragments.
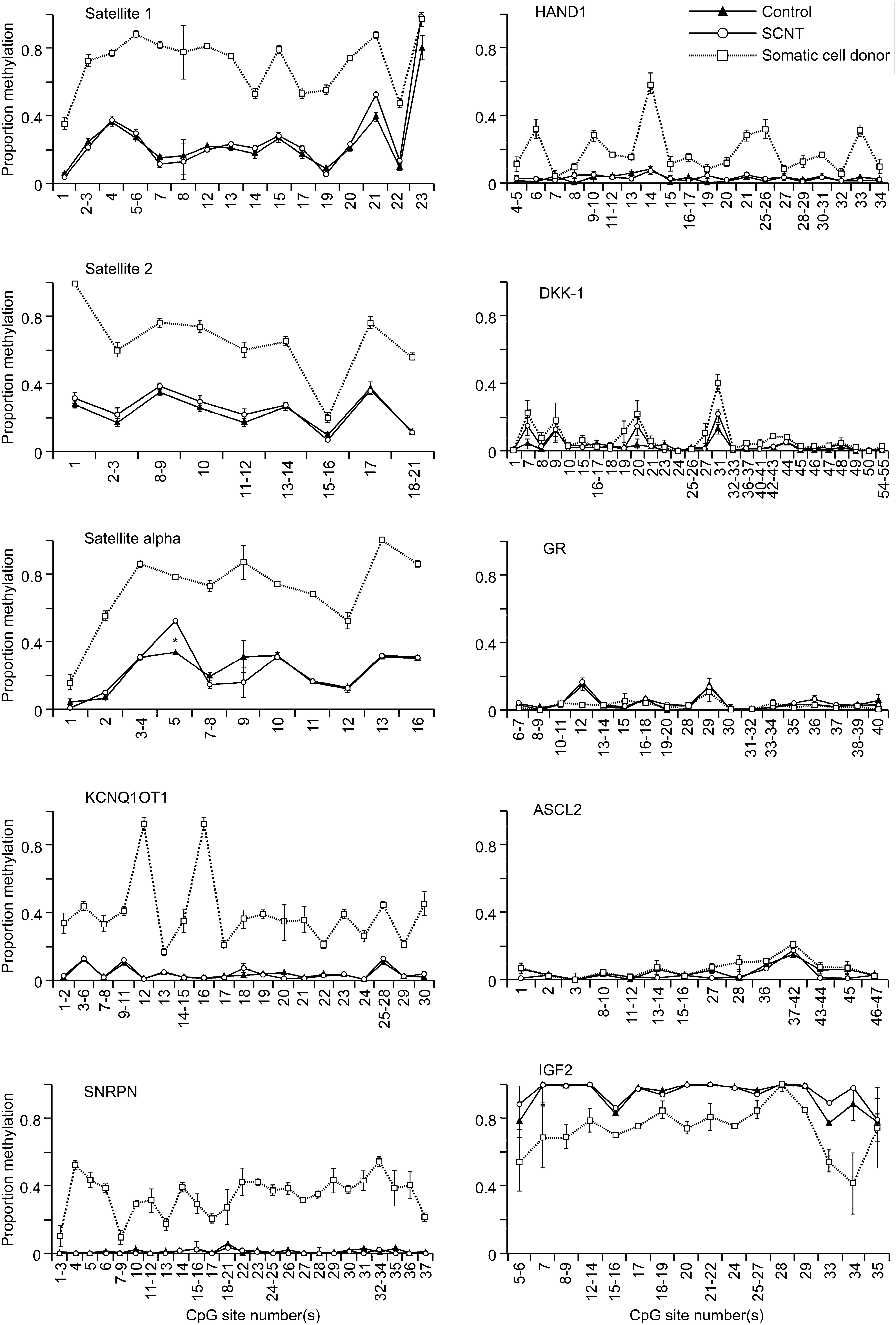
DNA methylation levels at CpG sites in the amplicons. Each CpG site or groups of sites that could be analyzed by Sequenom MassArray are arranged in the order that they appear in the DNA sequence, 5′ to 3′ on the x-axis. Where there is more than one CpG site in a fragment, the numbered CpG sites are grouped together in one position on the x-axis and the proportion of methylation refers to the most methylated site (Sequenom EpiTYPER 1 software). Where different cleavage fragments possessed the same mass/charge ratio and could not be distinguished from each other, the proportion of methylation in each of these fragment groups is represented as an average value. The y-axis represents the proportion of methylation at specific CpG sites in the region analyzed. The error bars represent the SEM. *p < 0.01 in satellite alpha.
The table shows the total number of CpG sites present in each amplicon, the number of CpG sites that were analyzed and the CpG sites that were contained within fragments that could not be resolved because they had identical mass/charge ratios. At these sites in any amplicon, the proportion of methylation is the averaged values from all these fragments.
Individual CpG sites or groups of CpG sites in the donor cells were methylated to varying levels in each region, giving distinctive methylation profiles. Methylation levels of the CpG sites in different regions ranged from very low (<0.05, e.g., ASCL2 and GR), to very varied (e.g., KCNQ1OT1, HAND1, and satellites 2 and alpha), to relatively high (0.6–1 (%), IGF2, and satellite 1) where 1 is complete methylation.
DNA methylation profiles in SCNT sperm were very similar to that of sperm from control bulls in all regions examined, with 174/175 CpG sites/groups of sites examined showing no differences between the two groups. The only CpG site at which a significant difference in methylation was detected was at CpG 5 in the satellite alpha repeat sequence (Fig. 1(c), p < 0.001), where statistical significance was observed even with the more stringent Bonferoni test.
Previously, we showed that the DNA methylation levels at the SNRPN and KCNQ1OT1 sites in fetal tissues varied greatly at each of the CpG sites between individuals (Couldrey and Lee, 2010). In contrast, there was very little variation in methylation levels between individual sperm samples (both SCNT and controls) at both of these regions (Fig. 1(d and e). Further analysis of additional sperm samples from SCNT bulls derived from four other donor cell lines (donor cell line: A, n = 4; B, n = 3; C, n = 3; D, n = 4), showed almost identical DNA methylation patterns (data not shown), including the hypermethylation noted at CpG 5 in satellite alpha. In general, there was very little individual variation in methylation levels at almost all the CpG sites examined in sperm.
The DNA methylation levels measured in sperm DNA were significantly different at the majority of CpG sites compared with the methylation levels measured in the nuclear donor somatic cells (Fig. 1), with 108/175 CpG sites/groups of CpG sites being significantly different (p < 0.05); of these, 87/175 were highly significant (p < 0.001). Hypomethylation of sperm DNA, relative to donor cells, was observed in all the three satellite sequences analyzed as well as in SNRPN, KCNQ1OT1, and HAND1. Methylation levels at the DKK-1 region in the nuclear donor cells were considerably lower compared with the six regions listed above, so the decrease in methylation seen in the sperm at several of the CpG sites was less apparent. ASCL2 and GR were virtually unmethylated in donor cell and sperm samples. In contrast, DNA methylation levels at the IGF2 exon 10 DMR were high (>0.6) at most CpG sites analyzed in the donor cells and almost completely methylated at most CpG sites in sperm DNA, consistent with a previous report (Gebert et al., 2006).
Sperm DNA was highly resistant to enzymatic digestion by the methylation sensitive HpaII restriction enzyme resulting in only high molecular weight DNA fragments (Fig. 2). In contrast, this same genomic DNA sample was extensively digested with MspI, a methylation insensitive isoschizomer of HpaII, resulting in fragments of a wide range of molecular weights, as illustrated by a smear of DNA following agarose gel electrophoresis.
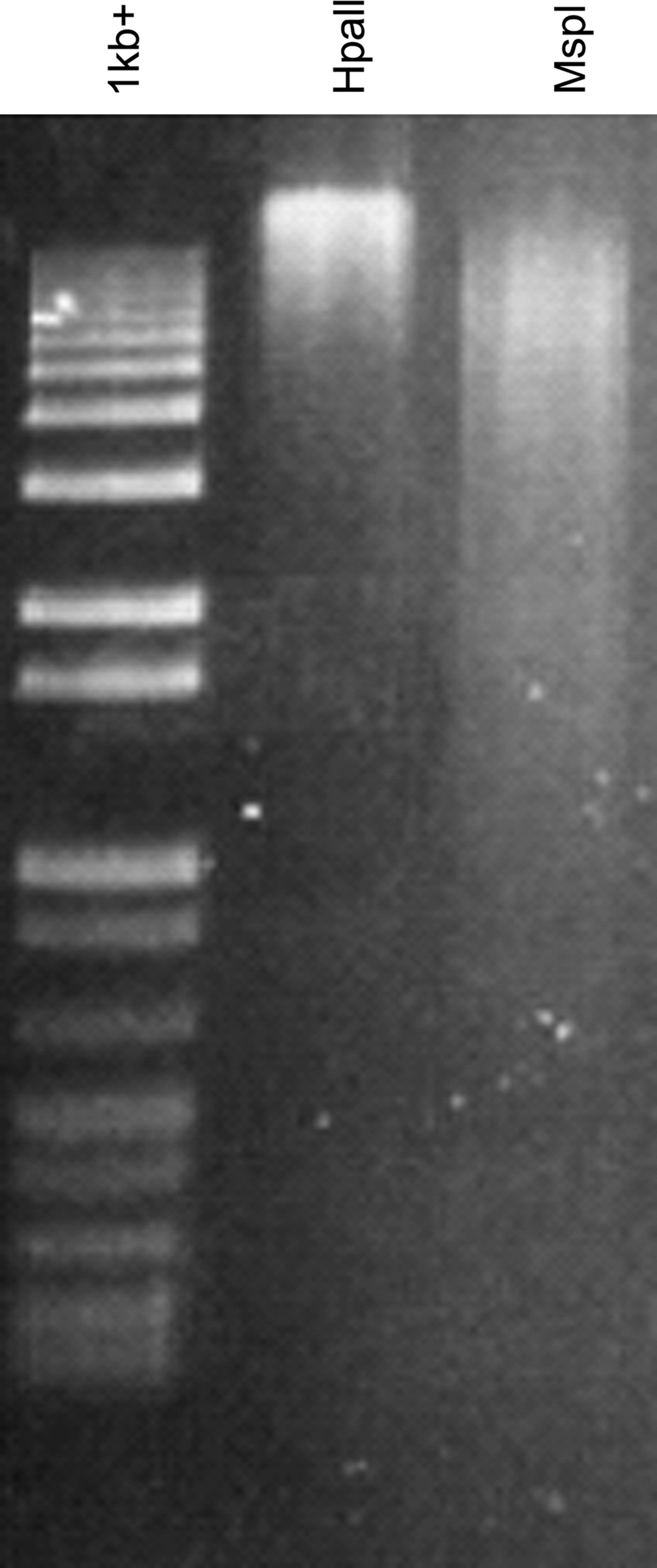
Methylation-sensitive and -insensitive restriction enzyme digestion of bovine sperm DNA. Genomic DNA isolated from washed control sperm was digested with HpaII or MspI restriction enzymes. A 1 kb+ DNA ladder (Invitrogen) was run alongside.
Discussion
We have examined the DNA methylation patterns at 10 genomic regions in sperm from SCNT bulls and compared them with control bull sperm of the same breed. With the exception of one site, which was significantly more methylated in sperm from SCNT bulls, the DNA methylation patterns in SCNT sperm were no different to control bull sperm. The one CpG site found to be different is located in the satellite alpha region; this site was also shown to be more highly methylated in SCNT fetal kidney tissues compared with the controls (Couldrey and Lee, 2010), although the difference between the SCNT and control samples was smaller in fetal tissues, compared with the difference observed in sperm samples. Given the degree of similarity in the methylation profiles between SCNT and control sperm for all the other genomic sites examined here, the biological significance of hypermethylation at this CpG site, which is in a noncoding region, is unclear. As this CpG site is present in multiple copies in each genome, and the measured DNA methylation levels are averaged over all these sites, the magnitude of the difference in methylation suggests that more of these sites must be methylated in satellite alpha in the DNA from SCNT sperm. This could either be due to more copies in individual cells becoming methylated, or more cells showing methylation in satellite alpha sequences. It is not possible to determine which of these explanations is leading to the increased methylation observed at this single CpG site.
A striking observation with DNA methylation patterns from sperm is the very low level of variation between individuals, even across different genetic backgrounds. Lack of variation between individuals could be explained by the homogeneity of the cell type in the sperm samples, that the genome is haploid or that there is tight regulation of methylation upon completion of sperm maturation. This tight regulation of methylation in gametes may indicate a low tolerance of variation in the epigenetic state of the gametes at the initiation of embryogenesis, as opposed to the epigenetic state of a somatic cell nucleus in a tissue, which can tolerate a degree of plasticity without serious consequence.
There was no difference in the methylation patterns at the SNRPN and KCNQ1OT1 sites between SCNT or control sperm samples, even though in several of the SCNT fetal tissues, DNA methylation patterns showed significant deviation from the pattern in control samples (Couldrey and Lee, 2010). This indicates that whatever epigenetic deviations were at these two sites in SCNT fetal tissues, the methylation patterns were correctly reset in the male germ cells on completion of spermatogenesis. The almost complete lack of methylation at these two regions in sperm, when compared with the level of methylation in the somatic donor cell or fetal tissue DNA, could be an indication that this is an imprinted methylation pattern for the male gamete in cattle. Both these sites are regarded in other species to be imprint control regions (Higashimoto et al., 2006; Horsthemke 1997).
The low levels of DNA methylation observed in sperm at both the repetitive satellite sequences and at all the single copy genes (except the IGF2 DMR) examined in this study was surprising, and appeared to contradict the dogma that sperm DNA is generally highly methylated. However, comparison of enzymatic digests of bovine sperm genomic DNA using either a methylation-sensitive or methylation-insensitive restriction enzyme clearly showed that the bovine sperm genome is indeed highly methylated.
That five of these seven single copy gene regions examined were virtually unmethylated in sperm could be explained by the fact that they are located in promoter regions, which tend to remain lowly methylated in most mammalian tissues (review in Illingworth and Bird, 2009). An alternative explanation comes from recent studies that have identified hypomethylation at developmentally important promoters in the sperm (Hammoud et al., 2009). However, neither of these possibilities accounts for the low levels of DNA methylation in the satellite sequences in sperm DNA relative to DNA from somatic cells or fetal tissues. These results illustrate just how the choice of which genomic region being investigated and the technique employed can lead to quite divergent viewpoints.
The difference in the methylation profiles between the nuclear donor somatic cells used for SCNT and the gametes from animals generated by SCNT, together with the observation that sperm from control and SCNT-derived bulls are virtually indistinguishable in their methylation profiles, demonstrates that the somatic cell genomes have been appropriately reprogrammed on passage through the germ line and that sperm from SCNT bulls are epigenetically no different compared with normal bull sperm. However, this kind of analysis can only measure mean methylation patterns in pooled sperm cells from any individual, so individual sperm cells with grossly abnormal epigenetic marks, if present at very low frequency, will not be detected. Therefore, we cannot exclude the possibility that there will be sperm cells with aberrant epigenetic marks, whether in SCNT or control bull samples. The chance of such a sperm with grossly abnormal epigenetic marks being able to fertilize an egg and drive embryo–fetal development to completion is very low.
In light of recent studies that have raised concerns about the potential for trans-generational passage of DNA methylation defects (Kobayashi et al., 2009; Lee et al., 2009; Stouder and Paoloni-Giacobino, 2009; Stouder et al., 2009), these results provide molecular evidence that accurate reprogramming is occurring and demethylation, together with both de novo and maintenance methylation is functioning correctly during gametogenesis in cloned bulls. This data also supports previous findings that semen parameters from cloned bulls are within normal ranges (Smith et al., 2007; Tecirlioglu et al., 2006), the fertility of cloned bulls is normal (Panarace et al., 2007; Tecirlioglu et al., 2006; Watanabe and Nagai, 2008, Wells and Lee, unpublished data), and that offspring of clones are generally normal (Wells et al., 2004).
In conclusion, these data provide molecular evidence that in at least 10 genomic regions examined, the correct epigenetic patterns have been established in the DNA of sperm from SCNT bulls after passage of the genome through the germline. This conclusion has significant agricultural implications for cloning as it indicates that any epigenetic aberrations that SCNT bulls may harbor in the somatic cells of the animal are unlikely to be passed onto their offspring through their gametes.
Footnotes
Acknowledgments
The authors thank Martin Berg for animal care and treatment, Tim Manley for the Sequenom MassARRAY sample analysis, Neil Cox for statistical assistance, and Pauline Hunt for graphics assistance. This work was supported by a grant from the New Zealand Foundation for Research, Science, and Technology (C10X0303).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.