
Abstract
Abstract
Although numerous mammalian species have been successfully cloned by somatic cell nuclear transfer (SCNT), little is known about gene expression of cloned pigs by SCNT. In the present study, expression profiles of 1-month-old cloned pigs generated from fetal fibroblasts (n = 5) were compared to those of age-matched controls (n = 5) using a 13K oligonucleotide microarray. The brain, kidney, and lung were chosen for microarray analysis to represent tissues from endoderm, mesoderm, and ectoderm in origin. In clones, 179 and 154 genes were differentially expressed in the kidney and the lung, respectively (fold change >2, p < 0.05, false discovery rate = 0.05), whereas only seven genes were differentially expressed in the brain of clones. Functional analysis of the differentially expressed genes revealed that they were enriched in diabetic nephropathy in the kidney, delayed alveologenesis as well as downregulated MAPK signaling pathways in the lung, which was accompanied with collapsed alveoli in the histological examination of the lung. To evaluate whether the gene expression anomalies are associated with changes in DNA methylation, global concentration of the methylated cytosine was measured in lung DNA by HPLC. Clones were significantly hypermethylated (5.72%) compared to the controls (4.13%). Bisulfite-pyrosequencing analyses of the promoter regions of differentially expressed genes, MYC and Period 1 (PER1), however, did not show any differences in the degree of DNA methylation between controls and clones. Together, these findings demonstrate that cloned pigs have altered gene expression that may potentially cause organ dysfunction.
Introduction
Materials and Methods
Tissue collection
Cloned pigs generated from fetal fibroblasts and control pigs were from the same genetically similar herd (Carroll et al., 2005). There were no clinical findings in clones until necropsy. Cloned pigs at 1 month of age (n = 5) had significantly lower mean body weight (6227.0 g) than that of genetically similar age-matched controls (n = 6) from conventional breeding (8263.3 g, p < 0.05). The relative mean weights (organ weight/100 g of body weight) of the liver and the spleen of clones, however, were significantly higher than those of controls (p < 0.05). To assess the overall degree of reprogramming, we chose the brain, kidney, and lung tissues to represent tissues of the endoderm, mesoderm, and ectoderm in origin for further study. All tissues were snap frozen in liquid nitrogen and stored at −80°C until use.
Microarray
Global gene expression analysis was conducted as described previously (Park et al., 2010). Briefly, total RNA was isolated from the collected tissues using the TRIzol reagent (Invitrogen, Carlsbad, CA), followed by DNase I (Invitrogen) treatment. After evaluation of integrity and purity, five qualified RNA samples from each tissue per group were used for further analyses. Reference RNA was prepared from tissues of naturally bred pig fetuses. Ten microgram of each RNA sample and reference RNA were reverse transcribed, labeled with Cy3 or Cy5 dye (GE Healthcare Life Sciences, Piscataway, NJ) for dye-swap, and hybridized to a porcine-specific microarray (Operon Technologies Inc., Alameda, CA) at 45°C for 20 h. The microarray contained 13,297 oligonucleotide probes that were designed from The Institute of Genomic Research (TIGR) Porcine Gene Index (SsGI release 5.0). After hybridization, signal intensities were extracted by GenePix Pro 6.0 (Molecular Devices, Union City, CA). Probes were flagged as present when more than 70% of their feature pixels were above two standard deviations (SD) of background in either Cy3 or Cy5 channel. The present probes were used for data analysis with GeneSpring 6.1 (Agilent Technologies, Palo Alto, CA). After Loess normalization (Clevel and Devlin, 1988) probes that were present in at least 90% of the microarrays with raw expression values >100, and an SD of <1.4 were designated as informative probes and used for further analysis (Jensen and Watt, 2006). Average linkage clustering by similarity measurement was performed (Eisen et al., 1998). Differentially expressed genes were identified by fold change (>2.0) and p-value cutoff ( = 0.05) followed by multiple test correction [False Discovery Rate (FDR) = 0.05]. Functional annotation and pathway analysis were performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (http://david.abcc.ncifcrf.gov) according to the user's guide (Dennis et al., 2003). Literature mining was conducted with PubMatrix (http://pubmatrix.grc.nia.nih.gov) to identify the biological and clinical relevance of the differentially expressed genes (Becker et al., 2003). Gene-to-gene and pathway-to-pathway interaction was investigated using PubGene (http://www.pubgene.org) by gene-to-gene cocitation networking (Jenssen et al., 2001).
Quantitative Real-Time RT-PCR (qPCR)
Expression levels of the representative four genes from the microarray were verified by qPCR with a 7500 Fast Real-Time PCR System (Applied Biosystems, Foster, CA). These genes included acyl-coA synthetase long-chain family 1 (ACSL1), CCAAT/enhancer binding protein alpha (CEBPA), notch homolog 3 (NOTCH3), and p38. Tubulin beta-2 (TBB2) was used as an endogenous control, and the reference RNA as a calibrator. Primers were designed using the Primer Express (Applied Biosystems) based on the microarray probe sequences from GenBank or SsGI release 12.0 (Table 1). The results were analyzed using Fast 7500 System SDS software 1.3.1 (Applied Biosystems) with comparative CT method.
Histology
The lung tissues of four cloned pigs and five controls were fixed in 10% neutral-buffered formalin and embedded in paraffin. Five-micron sections were cut and stained with hematoxylin and eosin (H&E). Structure of the alveoli was examined at a magnification of 200×.
DNA methylation analysis
Global and gene-specific concentrations of 5mC were measured as described previously (Park et al., 2010). Briefly, hydrophobic nucleosides from the genomic DNA of the lung were prepared by Nuclease P1 digestion (US Biological, Swampscott, MA) followed by Calf Intestinal Phosphatase dephosphorylation (New England Biolab, Ipswich, MA). 5-Methyl-2-deoxycytidine (5mC) standard was prepared by dephosphorylation of 5-methyl-2-deoxycytidine monophosphate (USB, Cleveland, OH). Other nucleoside standards were purchased from Sigma-Aldrich (St. Louis, MO). Global concentrations of 5mC were determined in duplicate by binary gradient reverse phase-HPLC, and the ratio between total dC (dC+5mC) and dG, and the percentage of 5mC [%5mC/(total dC)] were calculated from peak areas of individual nucleosides relative to standard curves. Differences between clones and controls were assessed using a one-sided Student t-test and an α-level of signification of p < 0.05. Candidate genes for gene-specific DNA methylation analyses were chosen based on four criteria: gene expression, availability of the promoter sequence information, abundance of CpG islands in the putative promoter regions, and literature evidence on gene regulation by DNA methylation. As a result, two genes (Myc and PER1) and their promoter regions were chosen for gene-specific DNA methylation analyses. PCR was performed under the optimized conditions as described elsewhere (Park et al., 2010) with bisulfite-treated lung DNA. Pyrosequencing reactions were conducted in a PyroMark MD system (Biotage, Charlottesville, VA) with PyroGold CDT reagents (Biotage). Quantification of DNA methylation was performed using PyroQ-CpG (Biotage), and one-sided Student's t-test was conducted for statistical analyses.
Results
Gene expression profiles in the brain
In the brain, 10,422 probes (78.3%) were identified as informative after filtration. Expression profiles of clones and controls clustered hierarchically into two distinctive groups with 0.917 of correlation coefficiency (r) for clones and r = 0.845 for controls, respectively. Statistical analysis revealed only seven differentially expressed genes in clones compared to controls (fold change >2.0, p < 0.05, FDR = 0.05). Among them the expression level of leukocyte cell-derived chemotoxin 2 (LECT2) was 2.15-fold higher in clones compared to controls. In contrast, six genes were downregulated in clones and these are procollagen-proline, 2-oxoglutarate-4-dioxygenase (P4HA3), CCAAT/enhancer binding protein delta (CEBPD), metallothionein 1A (MT1A), transgglutaminase 3 (TGM3), collagen type 1 alpha 1 (COL1A1), and pyruvate dehydrogenase kinase isoenzyme 4 (PDK4).
Gene expression profiles in the kidney
A total of 9,956 probes (74.8%) were identified as informative in the kidney. As in the brain, clones' expression profiles clustered together with r = 0.931, which were distinctly separated from controls of r = 0.678. Out of the informative probes, 179 genes were differently expressed (fold change >2, p < 0.05, FDR = 0.05) in clones compared to controls. Of these 49 were upregulated and 130 genes were downregulated in clones. Differentially expressed genes clustered into four groups of 30, 32, 92, and 24 genes, respectively (Supplementary Table 1; Supplementary Data are available online at http://www.libertonline.com/cell), and they were functionally enriched in “ion binding,” “cellular carbohydrate metabolism,” “porter activity” in group 1, “response to stress,” “cell differentiation,” “cell proliferation” in group 2, “response to stress,” “RNA Polymerase II transcription factor activity,” “morphogenesis” in group 3, and “extracellular region,” “acute-phase response” in group 4. In pathway analysis, the “adipocytokine signaling pathway” was identified to be dysregulated in clones. Literature mining showed that six dysregulated genes could lead to diabetic nephropathy in clones. Those included carbonic anhydrase 2 (CA2), glucose-6-phosphatase (G6PC), group-specific component (GC), inhibitor of kappa light polypeptide gene enhancer (IKBKB), pyruvate dehydrogenase kinase 2 (PDK2), and parathyroid hormone receptor 1 (PTHR1) (Table 2).
Gene expression profiles in the lung
In the lung, hierarchical clustering with 9955 informative probes (74.8%) revealed that clones and controls distinctly clustered with r = 0.943 and 0.903, respectively (Fig. 1A). Among these informative probes, 154 genes were differently expressed in clones (fold change >2, p < 0.05, FDR = 0.05) with 49 upregulated and 105 downregulated genes in clones. Differentially expressed genes were clustered into four groups (Fig. 1B, Supplementary Table 2), and the genes in group 1 enriched in “development,” group 2 in “plasma membrane,” group 3 in “response to stress,” “lipid metabolism,” and group 4 in “response to stress,” “morphogenesis,” “cell proliferation,” and “transcription from RNA Polymerase II promoter.” “MAPK signaling pathway” and “adipocytokine signaling pathways” were recognized to be dysregulated in clones as well. Eight genes associated with delayed alveologenesis were identified by literature mining, and they included ACSL1, ACSL4, aldehyde dehydrogenase 1 famliy member A3 (ALDH1A3), arginase 1 (ARG1), chromosome 9 open reading frame 13 (C5ORF13), CEBPA, complement factor B (CFB), integrin alpha 2 (ITGA2), mesenchyme homeobox 2 (MEOX2), NOTCH3, and superoxide dismutase 2 (SOD2) (Table 1). In order to study the interactions of the differentially expressed genes, we conducted cocitation and direct neighbor analyses. If two genes/pathways are cocited in the same study, it is likely that they may interact. This is a method of throughput analysis on gene/pathway interactions. Sixteen out of 20 delayed alveologenesis and MAPK pathway-associated genes linked together in a network as direct neighbor with maximum depth = 1, the most stringent criterion for gene interaction, having a range of 1 to 1517 of cocitations. Consistent with the observations in gene expression, histological examination of the lungs revealed that all of the lungs of cloned pigs showed mild atelectasis (collapsed alveoli) and only one out of five lungs of controls was identified with the same finding. Although there were no structural differences in the epithelium lining of airways or alveoli between clones and age-matched controls, the incidence of atelectasis was significantly higher (p < 0.01) in clones than in controls (Fig. 2 and Table 3).
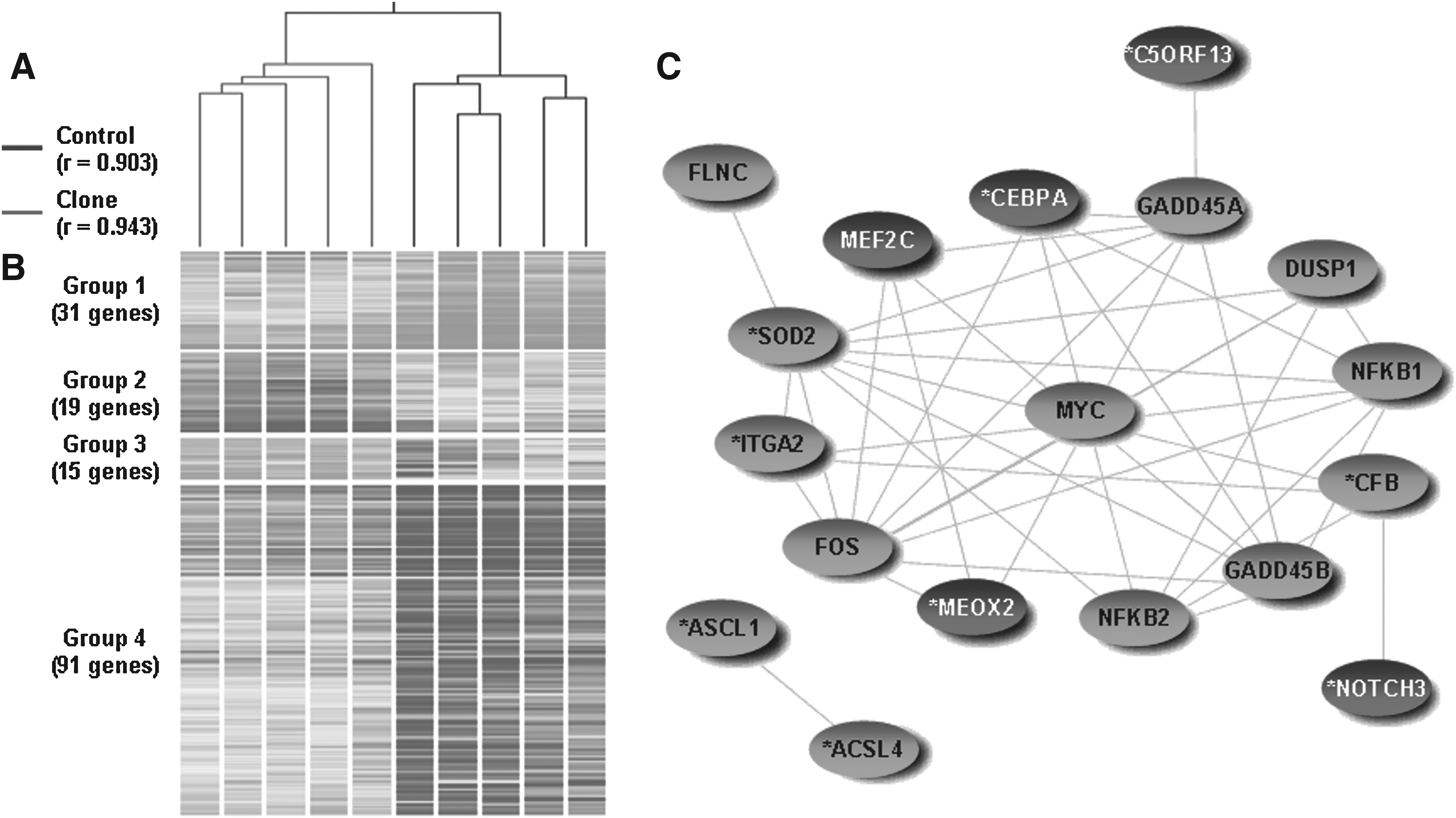
Gene expression analysis in the lung of 1-month-old cloned pigs. (

Histological examination of the lungs. (
p < 0.01, one-sided chi-square test.
Cocitations of other gene pairs in the lungs such as CEBPA2–FOS, CEBPA–MYC, SOD2–FOS, SOD2–MYC, ITGA2–FOS, and MEOX2–MYC were 10, 11, 10, 15, 3, and 2, respectively, were also found in the literature, suggesting that these genes collaborate in exerting their functions (Fig. 1C).
qPCR verification
Gene expression levels measured by microarray were consistent with those of qPCR (Table 4). ACSL1, a representative downregulated gene in clones, had 0.16-fold change in clones when compared with controls by Qpcr, and it was comparable to the 0.31-fold change by microarray. CEBPA and NOTCH3, representing upregulated genes in clones, showed 2.29- and 1.92-fold changes by qPCR, respectively. These corresponded with the 2.5- and 2.04-fold changes of CEBPA and NOTCH3 by microarray, respectively. p38 was one of the nondifferentially expressed genes between clones and control by microarray (fold change = 1.06), and qPCR also showed an expression ratio of 0.91.
p < 0.001, two-sided t-test.
DNA methylation analyses
As shown in Table 5, the ratios between total dC and dG ranged from 1.18 to 1.24 in both the control and clone groups, indicating that the method did not cause bias in the quantification of nucleosides. The mean proportions of 5mC to total dC were 5.72 ± 0.44% in clones and 4.13 ± 0.17% in controls of lung DNA (p < 0.001). The hypermethylation in clones occurred concurrently with upregulation of DNA methyltransferase 3 alpha (DNMT3a) in these animals.
p < 0.001, one-sided t-test.
Results on DNA methylation of specific genes are shown in Figure 3. Two genes that were differentially expressed between controls and clones were chosen: MYC, 2.1-fold downregulated in clones compared to controls (p < 0.01), PER1 was 2.8-fold downregulated in clones (p < 0.001). These genes have also been shown to be subjected to methylation regulation in the rat/human and the promoter sequences of these genes were available. Both were significantly and reversely related to the level of expression of DNMT3a. However, putative CpG islands in the promoter regions of both MYC and PER1 were hypomethylated (<4.93%). This is not statistically different than controls. To verify that our bisulfite-pyrosequencing method did not generate a biased hypomethylation status of the genes, we analyzed the putative CpG island of a ubiquitously expressed gene, RDBP, which is expected to be hypermethylated, and it was confirmed to have hypermethylation (>79.71%) in both groups (data not shown).
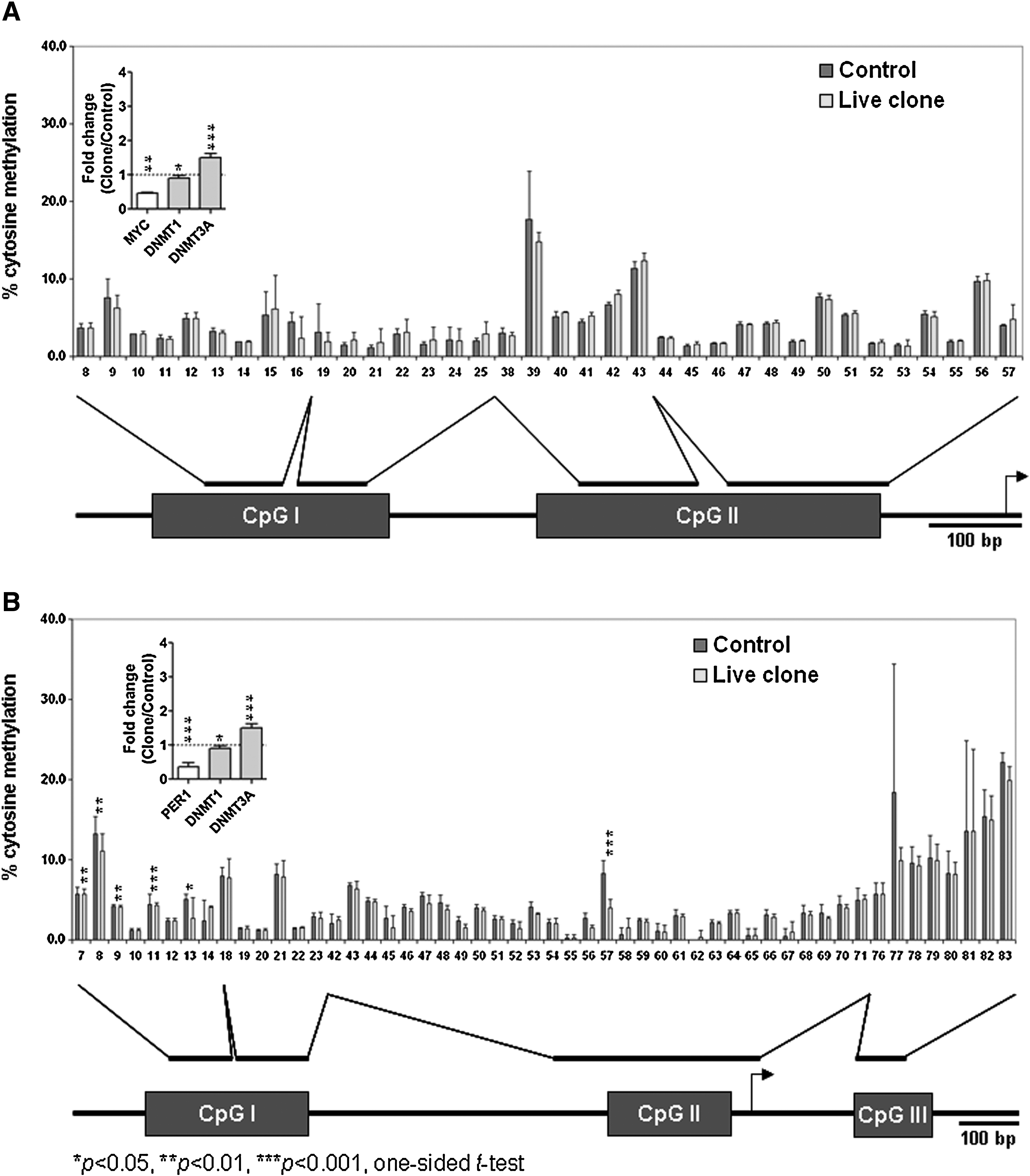
Gene-specific DNA methylation analysis of (
Discussion
In this study, we compared the global gene expression profiles and epigenetic status of 1-month-old cloned pigs to their conventionally bred counterparts and correlated these observations to clinical and pathological findings in these pigs. To our knowledge, this is the first description of the transcriptome and epigenetic status in healthy cloned pigs.
Even though the pigs we used here included both males and females, the samples were clustered into clones and controls, sex differences did not cause clustering separations, suggesting that they had little, if any, effect on expression profiles. Furthermore, the global gene expression profiles of the brain, kidney, and lung tissues of cloned pigs were less variable than those of age-matched controls, as would be expected. This is in contrast to findings in deceased neonatal cloned pigs whose kidneys and lungs had lower similarity in the expression profiles than controls (Park et al., 2010). The discrepancy could be due to the fact that deaths in newborn clones resulted from different causes, and gene expression in turn, may be more variable, even for cloned animals. To our surprise, many genes were found differentially expressed in the kidney and the lung of living, and apparent healthy clones compared to controls. These findings suggest that reprogramming errors can accumulate and persist throughout development and cloned animals can tolerate a wide range of abnormal expression of nonessential genes. Similar observations have also been reported in cloned mice in which normal-appearing clones had a large number of affected genes (Kohda et al., 2005).
Functional annotation and intensive literature mining revealed the clinical relevance of differentially expressed genes in organs of the cloned pigs. Six genes dysregulated in the kidney of cloned pigs were identified to have potential role in diabetic nephropathy: CA2 catalyzes the buffering of intracellular hydroxyl ions by CO2 in S1, S2, and S3 proximal segments of the nephrons, and renal adaptation to the metabolic acidosis potentially increases its expression (Tsuruoka et al., 1998). The strong induction of renal G6PC during starvation has been suggested to be responsible for glucose production in the kidney during long-term fasting (Simonnet, 1999). Furthermore, starvation upregulates PDK4 in parallel with PDK2 particularly in the gluconeogeneic tissues like liver and kidney (Sugden and Holness, 2002).
Among the dysregulated genes in the lungs of the cloned pigs, 11 were identified to have effects on delayed alveologenesis. For example, constitutive expression of NOTCH3 in the peripheral epithelium in the developing lung results in altered lung morphology and delayed development (Dang et al., 2003). MEOX2/homeobox B5 (HOXB5) expression is altered in hypoplastic lungs in nitrofen exposed mouse fetuses, and the persistent expression of MEOX2 in later stages of development and at birth causes delayed development and maturation of the lung (Chinoy et al., 2002). Lungs of mice with ectopically expressed CEBPA are of normal size but exhibit a phenotype characterized by fewer and larger developing epithelial tubules because of the disruption of branching process (Berg et al., 2006). Furthermore, synthesis of pulmonary surfactant is dependent upon CEBPA expression (Martis et al., 2006; Zhang et al., 2004). ALDH1 is located in the bronchial epithelium and alveolar parenchyma, and its temporal and spatial expression is associated with alveolar septation and postnatal lung development (Hind et al., 2002). The epithelial expression of ITGA2 is restricted to branch tips, and its spatial and temporal expression is associated with high level of expression of collagen mRNA (Coraux et al., 1998; Wu and Santoro, 1996), which is necessary for branching to occur normally. SOD2 null mutant mice show abnormalities in saccules as a result of delayed postnatal lung development (Asikainen et al., 2002; Tsan et al., 1998). Delayed alveologenesis predicted from gene expression analysis in this study was confirmed with histological examination of the lungs of the clones. Moreover, clinical observations were also made that were indicative of neurological disorders and respiratory problems in adult cloned pigs (R.S. Prather, unpublished data).
It was surprising that the MAPK kinase pathway, which is involved in many important physiological functions, was dysregulated in cloned pigs in both the kidney and lung. Previously we observed that dysregulated MAPK pathway was a prominent cause of pathogenesis in the kidney and the lung of deceased neonatal cloned pigs (Park et al., 2010). This common observation from both neonatal and 1-month-old cloned pigs suggests a consistent error by nuclear transfer in pigs. Higher activity of the MAPK is important for the survival of cloned pig embryos. However, this higher levels of MAPK needs to be fine tuned to a normal level during fetal development. Failure to achieve this by the cloned animals affects their survivability and health status. It is known that disruption of MAPK pathway was associated with pathogenesis in the brain and the kidney (Haneda et al., 2001; Lee et al., 2006; Toyoda et al., 2004). Although it is not clear that altered MAPK pathway could delay the alveologenesis process, differentially expressed genes that could cause delayed alveologenesis were closely related to the MAPK pathway as they were identified in the same network in this study. Together, these results imply that adjustment in the activity of the MAPK kinase should be an important area of study in pig cloning.
In contrast to less than 2% of 5mC composition in several eukaryotic DNA from calf, salmon, and humans, pigs have been reported to have more than 5% of 5mC in DNA (Kuo et al., 1980), and is compatible to the global concentration of 5mC measured in this study. This may be due to the higher proportions of 5mC and GC content, as well as more CpG islands in the pig than the other species (Amarger et al., 2002). Cloned pigs at 1 month of age had a higher concentration of 5mC in their lung DNA than that of age-matched controls. It may suggest a hypermethylated genome in clones when compared with their conventionally produced counterparts. The observation of the hypermethylated genome was coincident with the mechanism of gene silencing through DNMT3a-mediated DNA methylation, which was upregulated in clones. It is noteworthy that considerably more genes were downregulated in cloned pigs than controls. Again, this could be caused by hypermethylation and upregulation of DNMT activity in clones. In contrast, an inverse correlation was observed between the global concentration of DNA methylation and the number of differentially upregulated genes in the deceased neonatal cloned pig, concordant with the mechanism of gene silencing by DNA hypermethylation (Park et al., 2010). Although it is uncertain what level of epigenetic modification of DNA is needed for successful SCNT, DNA methylation may be related to the survivability and well-being of cloned animals.
Gene-specific DNA methylation analyses revealed that cloned pigs had similar degree of DNA methylation as their controls. The genes studied herein were differentially expressed in clones when compared with controls, and their expression has been reported to be regulated by DNA methylation at their promoter regions in rats and humans (Tsujiuchi et al., 1999; Yeh et al., 2005). It cannot be excluded that, in contrast to cloned cows, the cloned donor genome of pigs undergoes typical demethylation processes, the patterns of which are similar to their counterparts (Kang et al., 2001). The lack of differences in the degree of gene-specific DNA methylation might be caused by the heterogeneous population of the tissue, which causes the dilution effect to interfere with precise quantification of DNA methylation levels. Nonetheless, it is noteworthy that there are species-specific differences in modifying the epigenetic status of cloned donor genomes, implying that the differentially expressed genes investigated in this study may be regulated by a mechanism other than DNA methylation in pigs.
In summary, this study demonstrates that cloned pigs studied at 1 month of age may not be severely affected animals in regard to their health, but harbor magnified abnormalities from inadequate nuclear reprogramming. Live-cloned pigs can tolerate significant gene expression dysregulation.
Footnotes
Acknowledgments
This work was supported by Grant 1265-31000-091-02S from the United States Department of Agriculture Agricultural Research Service (USDA-ARS) and from the National Institutes of Health (RR013438).
Author Disclosure Statement
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.