
Abstract
Abstract
The development of embryos produced by somatic cell nuclear transfer (SCNT) using vitrified oocytes as cytoplast recipients has been reported in cattle but not in sheep. This study investigated the parthenogenetic development of ovine oocytes vitrified and thawed at the germinal vesicle (GV) stage, matured in vitro, and then activated using two activation protocols. The optimal activation protocol was then used to assess development when vitrified oocytes were used as cytoplast recipients for SCNT. No blastocysts were obtained from vitrified oocytes activated by CA+CHX/CB (calcium ionophore A23187 + cycloheximide, and cytochalasin B); in contrast, vitrified oocytes activated by Sr/CB (strontium chloride (SrCl2) + cytochalasin B) developed to blastocyst, although the number was significantly lower (p < 0.05) than in toxicity and control groups (3.8 vs. 20.0 and 27.3%, respectively). In SCNT embryos, cleavage at both 24 and 48 h postactivation (31.0 vs. 55.1% and 48.0 vs. 85.0%) was significantly lower (p < 0.05) in vitrified oocytes compared to controls. However, no significant differences were observed in the frequency of development to blastocyst (13.0 vs. 23.4%), the number of hatched blastocysts (7.0 vs. 10.3%), total cell numbers (90.3 ± 4.9 vs. 97.6 ± 4.6), number of apoptotic nuclei (13.1 ± 0.9 vs. 13.2 ± 1.4), or the proportion of diploid embryos (60.0 vs. 75.0%). This study demonstrates for the first time that ovine oocytes vitrified at the GV stage can be used successfully as recipient cytoplasts for SCNT.
Introduction
We have previously reported that GV stage ovine oocytes can be vitrified and subsequently thawed and matured in vitro with high survival and maturation rates (Moawad and Campbell, 2008; Moawad et al., 2010). In the present study parthenogenetic development was compared following two activation protocols; (1) a combination of calcium ionophore A23187, cycloheximide, and cytochalasin B (CA+CHX/CB), and (2) strontium and CB (Sr/CB). The optimal activation protocol was then used to determine the developmental competence of SCNT embryos produced using ovine oocytes vitrified at the germinal vesicle stage and subsequently matured and enucleated as cytoplast recipients.
Materials and Methods
All chemicals and reagents were purchased from Sigma-Aldrich (Dorset, UK), unless otherwise stated.
Oocyte collection
Ovine ovaries collected at a local slaughterhouse were placed into phosphate-buffered saline (PBS) at 25°C in a thermos flask and transported to the laboratory within 3 h. Follicles 2–3 mm in diameter were aspirated using a 21-gauge needle attached to 10-mL syringe and the follicular material placed into 50-mL conical tubes in a warm box at 39°C. After settling, the upper follicular fluid was discarded and the remaining material diluted with oocyte washing medium [HEPES-buffered TCM 199 (H-TCM 199); Gibco, Paisley, Renfrewshire, UK, supplemented with 10% fetal bovine serum (FBS); Gibco] and transferred into a 95-mm Petri dish. The Petri dish was transferred to a heated stage on a dissecting microscope (Leica MZ125, Leica, Wetzlar, Germany). Cumulus–oocyte complexes (COCs) with at least two to three compact layers of cumulus cells and a homogeneous cytoplasm were selected and transferred into fresh washing medium at 39°C for processing.
Vitrification and warming of COCs
Selected COCs were washed three times in a base medium (BM) (H-TCM 199 supplemented with 10% heat-inactivated FBS) and then placed into 500 μL drops of equilibration solution [10% ethylene glycol (EG) plus 0.25 M trehalose in BM] for 3 min on a warm stage at 39°C. After equilibration the oocytes were placed into 20 μL drops of vitrification solution [20% EG and 20% dimethylsulfoxide (DMSO)] at 39°C and then either vitrified or processed through the warming solutions (toxicity control). For vitrification, a thin film of vitrification solution was obtained by dipping the cryoloop into the vitrification solution. Three to five oocytes were then suspended on the film using a fine pulled-glass mouth pipette and the cryoloop immediately plunged into a cryovial filled with liquid N2 (LN2) and then the vial sealed and transferred into LN2 for storage (Lane et al., 1999). Following oocyte equilibration, the vitrification procedure was completed within 1 min. For thawing and warming of the vitrified oocytes, the cryovial which remained submerged in LN2 was opened, the cryoloop removed and placed directly into 500 μL BM containing 10% EG and 1 M trehalose. The cryoloop was removed and the oocytes maintained in this solution for 3 min and then transferred into 500 μL of 0.5 trehalose solution in BM for 3 min followed by 500 μL of BM for another 3 min. All thawing procedures were carried out in four-well dishes (Nunc, Roskilde, Denmark) at 39°C on a heated stage.
In vitro maturation of ovine oocytes
Oocytes were matured in vitro as previously described (Lee and Campbell, 2006). Briefly, morphologically normal COCs selected before or after vitrification and thawing, were washed twice in maturation medium (bicarbonate-buffered TCM 199 with Earle's salts; Gibco) supplemented with 10% FBS, 5 μg/mL FSH (Vetropharm, Ireland), 5 μg/mL LH (Vetropharm), 1 μg/mL estradiol-17β, 0.3 mM sodium pyruvate, 100 μM cysteamine, and 50 μg/mL gentamycin. Groups of 40–45 oocytes were transferred into 500 μL of prewarmed maturation medium overlaid with 300 μL mineral oil in four-well dishes (Nunc) and incubated for 24 h at 39°C in a humidified atmosphere of 5% CO2 in air.
Parthenogenetic activation
At 24 hpm (hours postonset of maturation), cumulus cells were removed from the oocytes by repeated pipetting of COCs in H–TCM 199/PVP containing 300 IU/mL hyaluronidase. Oocytes were selected based on the presence of a visible first polar body (PBI) and homogenous cytoplasm and activated using one of two protocols: (1) groups of oocytes were incubated in 5 μM calcium ionophore (A 23187) in HEPES-buffered Synthetic Oviduct Fluid supplemented with 4 mg/mL BSA (H-SOF-BSA) for 5 min at 39°C. Oocytes were then washed twice in H-SOF-BSA and once in embryo culture medium (mSOFaaciBSA, modified SOF medium containing 2% (v/v) BME-essential amino acids, 1% (v/v) MEM-nonessential amino acids, myoinositol, sodium citrate, and fatty acid free-BSA 4 mg/mL), and then cultured in mSOFaaciBSA supplemented with 10 μg/mL CHX and 7.5 μg/mL CB for 4–5 h at 39°C in a humidified atmosphere of 5% CO2 (CA+CHX/CB). (2) selected oocytes were washed three times in 500 μL of calcium free CZB (Chatot, Ziomet, and Bavister) medium and then incubated in calcium free CZB medium containing 10 mM SrCl2 (strontium chloride) and 7.5 μg/mL CB for 4–5 h at 39°C in a humidified atmosphere of 5% CO2 (Sr/CB). The calcium-free CZB was prewarmed in the incubator for at least 1 h before adding 100 × strontium solution (10 μL/mL) in order to avoid strontium precipitation.
SCNT
All SCNT procedures were carried out as previously described (Lee and Campbell, 2008). Briefly, at 15–16 h after the onset of maturation (hpm) oocytes were denuded of cumulus cells and examined for the presence of an extrusion cone. Selected oocytes were cultured in H-SOFBSA supplemented with Hoechst 33342 (5 μg/mL) for 15 min at 39°C. Groups of 15–20 oocytes were then transferred into 50 μL drops of manipulation medium [H-SOF supplemented with 4 mg/mL BSA and 7.5 mg/mL cytochalasin B (CB)] under oil in a 90-mm petri dish at 39°C. For enucleation the zona pellucida was cut using a single pulse from a XYclone laser (Hamilton Thorne, Beverly, MA, USA), a small portion of cytoplasm was then aspirated from the extrusion cone using a 20-μm glass pipette. Enucleation was confirmed by examining the aspirated karyoplast by fluorescence microscopy and successfully enucleated oocytes placed back into the maturation medium. At 22–24 hpm enucleated oocytes were returned to the manipulation chamber, a single donor cell was placed under the zona pellucida of each enucleated oocyte. For fusion, two to three couplets were placed between the electrodes in 200 μL of 0.3 M mannitol in a fusion chamber (Eppendorf, Germany), manually aligned, and then exposed to two DC pulses of 1.25 kV/cm for 30 μsec using a multiporator (Eppendorf). The couplets were then placed into 50 μL drops of mSOFaaciBSA at 39°C in a humidified atmosphere of 5% O2, 5% CO2, 90% N2 for 1 h and then checked for fusion. Fused couplets were washed three times in 500 μL of calcium-free CZB medium at 39°C and then activated using Sr/CB as described above.
In vitro culture and evaluation of parthenotes and SCNT embryos
At 4–5 h postactivation (hpa) the reconstructed embryos were washed three times in H-SOF-BSA and twice in embryo culture medium. Groups of 10–15 embryos were cultured in 50 μL drops of mSOFaaciBSA under mineral oil at 39°C in a humidified atmosphere of 5% O2, 5% CO2, and 90 % N2. At 24 and 48 hpa cleaved embryos were transferred to fresh culture medium. Development to morula and blastocyst stages was assessed at day 5 and day 7 postactivation, respectively. Blastocyst stage embryos were morphologically evaluated under a stereomicroscope; they were then stained with 10 μg/mL Hoechst 33342 and total cell numbers determined by epifluorescence (Leica DMIRB, Germany).
Apoptosis assay
On day 7 of culture, blastocysts produced following parthenogenetic activation of oocytes or SCNT were selected and washed three times in 200 μL drops of PBS/PVP. Washed blastocysts were then incubated in 4% PFA containing 40 μg/mL Hoechst 33342 for 30 min at room temperature, followed by two washes in 200 μL PBS/PVP. Fixed embryos were transferrred into 500 μL of freshly prepared permeabilization solution [0.1% (v/v) Triton X-100 in PBS/PVP], incubated for 5 min at room temperature, and then washed twice in 200 μL PBS/PVP. DNA degradation was determined by Terminal Transferase dUTP Nick End Labeling (TUNEL) using an in situ cell death detection kit (Roche Diagnostics, Mannheim, Germany) according to manufacturer's instruction. Briefly, the embryos were incubated in 50 μL of the TUNEL reaction mixture (1:10 dilution terminal deoxynucleotidyl transferase in label solution) at 39°C for 45 min in a humidified chamber; they were then washed in 200 μL PBS/PVP, and mounted under coverslips on glass slides in Vectashield containing DAPI (Vector Laboratories Inc., Burlingame, CA, USA). DNA fragmentation was visualized using an epifluorescence microscope (Leica DMR, Germany) fitted with a digital camera (Hammamatsu, Japan) and image analysis software (Simple PCI, Compix Inc., USA).
Cytogenetic analysis of SCNT produced blastocysts
Day 7 blastocysts produced by SCNT of vitrified and control oocytes were prepared for karyotype analysis as previously described (King et al., 1979) with minor modifications. Dividing cells within the blastocysts were arrested at mitotic metaphase by incubating the embryos in culture medium containing 0.05 μg/mL demecolcine solution for 3 h at 39°C in a humidified atmosphere of 5% O2, 5% CO2, and 90% N2. They were then washed once in PBS/PVP, transferred to 0.8% sodium citrate for 10 min at room temperature, followed by 10 min in 0.56% potassium chloride. The blastocysts were then fixed in a solution consisting of 1:1.2 sodium citrate (0.8%) and methanol:acetic acid (2:1) for 15 min at room temperature and then mounted individually on freshly cleaned glass slides in a minimal volume of fixative. Immediately after mounting each slide was observed using a stereomicroscope (Leica), prior to drying of the fixative small volumes of freshly prepared methanol:acetic acid (1:1) were dropped onto the embryo until complete cell spreads were obtained. The spreads were then air dried, stained with 4% (v/v) Geimsa for 5 min, washed with water, and air dried at room temperature. Ploidy was evaluated using phase contrast microscopy (Leica DMR, Germany) at 100× magnification under oil immersion. Embryos were classified as being normal when the chromosome number was diploid (54, 2n) or abnormal when chromosome number was haploid (n) or polyploid (≥3 n).
Statistical analysis
At least three replicates were carried out for each experimental group. Development data following activation and SCNT were analyzed using chi-squared test. Total cell numbers, numbers of apoptotic nuclei and apoptotic indexes were analyzed using unpaired student's t-test (http://www.graphpad.com). All results are considered to be statistically significant at p < 0.05.
Results
Effects of activation protocol on parthenogenetic development of ovine oocytes
Cleavage and subsequent development of ovine oocytes vitrified at the GV stage and activated by two different methods are presented in Table 1. The frequency of cleavage was significantly (p < 0.05) lower in vitrified oocytes activated by either protocol (CA+CHX/CB or Sr/CB) at both 24 (6.2 and 3.8%) and 48 hpa (28.4 and 27.5%) compared to toxicity and control groups. Irrespective of the method of activation, no significant differences were observed in the frequency of development to the morula stage between vitrified and toxicity groups (range: 18.8 to 35.7%). However, vitrification of oocytes resulted in a significantly (p < 0.05) lower frequency of development to morula and blastocyst stages following activation by either protocol compared to control groups. Interestingly, only activation by Sr/CB was able to support development to blastocyst (3.8%) of vitrified oocytes, although this was significantly lower compared to controls (p < 0.05) (Table 1). In comparing development of those oocytes that had cleaved at 48 hpa, no significant differences were observed in the frequencies of development to blastocyst between vitrified (13.6%) or toxicity and control oocytes activated by CA+CHX/CB (25 and 21.6%). However, the numbers of cleaved embryos reaching the blastocyst stage was significantly lower in control oocytes activated by CA+CHX/CB than those activated by Sr/CB (43.2%). No significant differences were observed in the proportions of hatched blastocysts between vitrified (1.3%) and other groups (4.3% to 7.1%) or in the total cell numbers (Table 1). No significant differences were observed in the number of apoptotic nuclei in blastocysts produced by Sr/CB activation between vitrified, toxicity, and control groups (15.8 ± 2.8, 10.6 ± 1.3, and 12.9 ± 0.7, respectively). The same trend was noticed in terms of apoptotic indexes (number of apoptotic nuclei / total cell numbers in blastocyst) with values of (17.8 ± 5.2, 12.1 ± 0.9, and 15.1 ± 1.2 in vitrified, toxicity, and control groups, respectively).
Ovine oocytes vitrified and exposed to vitrification solution (toxicity control) were matured for 24 h. Oocytes exhibiting a first polar body were activated using 5 μM calcium ionophore (CA, A23187) in H-SOF-BSA for 5 min and then transferred to mSOFacciBSA medium supplemented with 10 μg/mL cycloheximide (CHX) and 7.5 μg/mL cytochalasin B (CB) for 4 to 5 h at 39°C in a humidified atmosphere of 5% CO2 (CA+CHX/CB). Alternatively, oocytes were activated by 10 mM SrCl2 (strontium chloride) and 7.5 μg/mL CB in calcium free CZB medium (Sr/CB). Cleavage rates were evaluated at 24 and 48 hpa (hours postactivation). Morula and blastocysts development as well as total cell numbers were also assessed at day 5 and day 7, respectively. Numbers of apoptotic nuclei and apoptotic indexes in day 7 blastocysts produced by Sr/CB activated oocytes were assessed using TUNEL. N/A means not applied.
Values with different superscripts in the same column are significantly different (p < 0.05).
Development of SCNT embryos
Ovine oocytes vitrified and thawed at the GV stage and subsequently matured in vitro were used as cytoplast recipients for SCNT. No differences in the frequencies of enucleation and fusion were observed between control and vitrified cytoplasts (Fig. 1 and Table 2). The number of cleaved embryos at both 24 and 48 hpa was significantly lower (p < 0.05) when using vitrified/thawed oocytes (31 and 48%) compared to control (55.1 and 85.0%). However, no significant difference was observed in the frequencies of development to morula (38.0 vs. 46.7%) and blastocyst stages (13.0 vs. 23.4%). Based on the numbers of embryos that had cleaved at 48 hpa, the same frequency of development to blastocyst was obtained in both groups (∼27%). A similar trend was observed in the proportion of hatched blastocysts (7.0 vs. 10.3%, in vitrified and control groups, respectively; Fig. 2A). No significant differences were observed in the total cell numbers, numbers of apoptotic nuclei, or apoptotic indexes between the two groups (Table 2 and Fig. 2B).
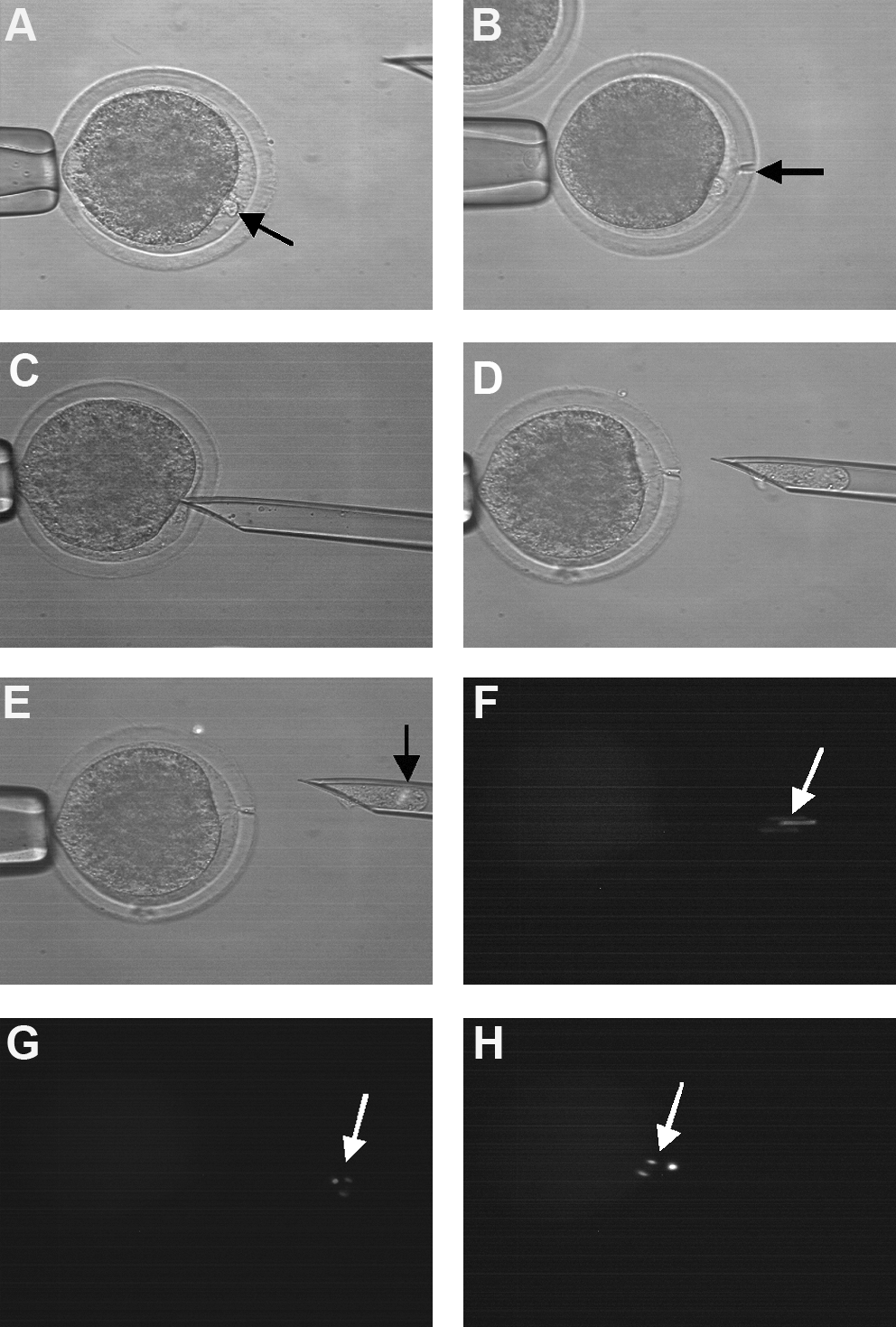
Enucleation of ovine oocytes vitrified at the GV stage. (
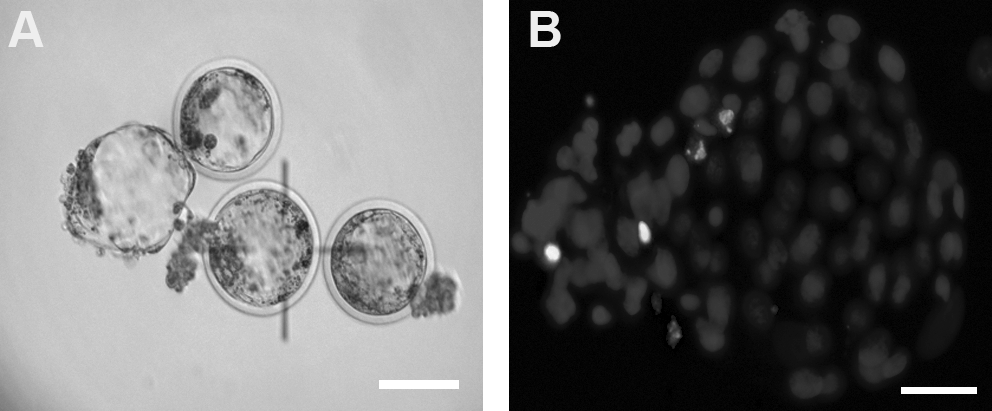
Day 7 blastocysts produced by SCNT using ovine oocytes vitrified at the GV stage. (
Vitrified GV oocytes were enucleated at 15–16 hpm then were cultured in maturation medium until cell fusion. Oocyte cell couplets in vitrified and fresh (control) groups were fused at 22 to 24 hpm. Fused couplets were then activated using Sr/CB activation method. Following activation, reconstructed embryos were cultured in mSOFaaciBSA for 7 days. Cleavage 24 and 48 hpa, morula (day 5 pa), and blastocyst (day 7 pa) development were evaluated. Total cell numbers and numbers of apoptotic nuclei were examined using Hoechst and TUNEL assay.
Values with different superscripts in the same column are significantly different (p < 0.05).
Karyotype of SCNT blastocysts
The ploidy of SCNT derived blastocysts was evaluated and the results are summarized in Table 3. Embryos were categorized as diploid (2n), haploid (n), or polyploid (≥3n); diploid embryos were considered as normal, others were reported as abnormal. Although the proportion of embryos with a normal diploid chromosome number (54, 2n) appeared lower in SCNT blastocysts obtained using vitrified oocytes compared to controls (60.0 vs. 75.0%, respectively) this was not statistically significant. In addition, no significant difference was observed in the numbers of haploid (20.0 vs. 10.0%) and polyploid (20.0 vs. 15.0%) embryos between the two groups.
Embryos were categorized as being normal when they contained a diploid karyotype (54, 2 n) or abnormal when haploid (n) or polyploid (≥3 n) chromosomes.
Discussion
This study demonstrates for the first time that vitrified/thawed GV stage ovine oocytes can be matured and support development when used as recipient cytoplasts for SCNT. The success of nuclear transfer depends on a range of interacting factors including efficient oocyte activation, which is a key step in the process (Campbell, 1999). Parthenogenetic activation and culture in vitro provides a good model for evaluating the developmental potential of cryopreserved oocytes (Morbeck et al., 2009). The present study compared the effectiveness of two activation protocols (1) CA+CHX/CB and (2) Sr/CB to promote parthenogenenetic development of vitrified/thawed ovine GV oocytes. In control oocytes both protocols supported development of ovine parthenotes; however, previous studies have suggested that activation of ovine oocytes with Sr/CB may improve developmental competency producing blastocysts with an increased total cell number and a reduction in the number of apoptotic cells (Choi and Campbell., 2008). In this study both treatments activated oocytes vitrified at the GV stage, and supported preimplantation development to the morula stage. However, the frequency of cleavage and development was significantly reduced compared to control and toxicity control oocytes. One explanation of these observations is that rapid freezing procedures may affect the permeability of the oolemma or other internal properties of oocytes that respond to calcium ionophore (CA) treatment. Therefore the concentration of CA and duration of exposure used for activation of freshly matured oocytes may not be optimal for vitrified oocytes. Further investigation is required to evaluate the effects of vitrification and thawing procedures on oocyte activation and subsequent development.
Previous studies on parthenogenetic activation have shown that the most effective protocols are those that promote a series of intracellular calcium oscillations (Vitullo and Ozil 1992). In mice, strontium has been reported to induce a series of intracellular calcium rises in both mature and immature oocytes and also in early embryos. Although the mechanisms by which strontium causes Ca2+ oscillations are unknown, it is mediated through IP3 (Inositol-1, 4, 5-triphosphate) receptors and requires activation of phospholipase C, which is similar to that seen in normal fertilization (Jellerette et al., 2000; Zhang et al., 2005). In sheep, previous studies have shown that treatment with 10 mM SrCl2 in combination with CA was more effective for activation of fresh MII oocytes when compared to other concentrations of SrCl2 or by activation using CA+CHX/CB (Choi and Campbell, 2008). Although blastocyst development from vitrified oocytes was only observed following activation with Sr/CB, both activation protocols resulted in the same proportions of blastocyst development in other groups (toxicity and control). The lack of toxicity associated with exposure to cryoprotectants observed in these studies may be due to the short exposure time employed during the vitrification procedure overcoming the detrimental effects associated with the high concentrations used.
In general freshly matured oocytes from in vitro or in vivo sources are used as cytoplast recipients for NT. The development of successful cryopreservation procedures would have significant benefits for laboratories performing nuclear transfer, oocytes could be collected and stored when available eliminating seasonal fluctuations in oocyte quality, animal availability, and dependence on other factors such as timing of slaughter and oocyte shipment. The present study evaluated the use of ovine oocytes vitrified at the GV stage as cytoplast recipients for SCNT. Although the frequencies of cleaved embryos at 24 and 48 hpa were significantly lower when using vitrified oocytes, development to morula and blastocyst stages did not differ significantly as compared before to controls (Table 2). These results indicate that vitrified GV stage ovine oocytes can be subsequently matured and used as recipient cytoplasts for SCNT with a high proportion developing to good quality blastocyst stage embryos. These results suggest that vitrification of immature GV stage ovine oocytes may provide opportunities for the establishment of oocyte cryobanks, allowing collection and storage of oocytes not only to facilitate SCNT but also from rare or endangered breeds or species. Previous studies have reported that the damaging effects of freezing–thawing were apparent only up to the two-cell stage; however, beyond this stage no further influences were observed on the development of cleaved embryos to morula or blastocyst stages (Schroeder et al., 1990). More recently, oocyte vitrification has been reported to reduce the level of maternal mRNAs in ovine oocytes (Succu et al., 2008), this may contribute to the lower cleavage observed following SCNT using vitrified oocytes. Differences that were observed in the frequencies of blastocyst development following parthenogenetic activation of vitrified oocytes compared to SCNT support previous studies that nuclear materials, surrounding microtubules and meiotic spindles are more adversely affected by cryopreservation (Dinnyes et al., 2000; Kubota et al., 1998; Moawad et al., 2010; Tominoga et al., 2005) than other cytoplasmic components; however, effects on other cytoplasmic components should not be neglected (Dinnyes et al., 2000).
The total cell number and the number of apoptotic nuclei in blastocyst stage embryos are considered as indicative of embryo quality (Choi and Campbell, 2010). In the present study, we obtained blastocyst embryos following SCNT with ovine oocytes vitrified at the GV stage, which had total cell numbers and apoptotic indices comparable to control cytoplasts. In contrast, previous studies in cattle and pigs reported lower cell numbers in blastocysts produced by SCNT and parthenogenetic activation of vitrified/thawed MII oocytes compared to control (Atabay et al., 2004; Somfai et al., 2006; Tominaga et al., 2005). Together, these observations suggest that oocytes vitrified at the GV stage and subsequently matured may undergo less damage than those cryopreserved at MII and support improved development when used as cytoplast recipients for SCNT. Previous studies reported that in subsequent culture cryopreserved oocytes often degenerated via apoptosis (Men et al., 2003); however, addition of insulin-like growth factor 1 inhibited this (Makarevich and Markkula, 2002). It has been suggested that the improved development of embryos observed using oocytes that had been vitrified and handled in media containing FBS may be due to the presence of growth factors in the FBS preventing apoptosis (Horvath and Seidle, 2008). The similarities in developmental potential reported here following SCNT of vitrified oocytes compared to control may be due to several factors including; the vitrification protocol, cryoprotectant combinations, or the activation method. Also, the stage at which enucleation was carried out (AI-TI) may influence blastocyst development in vitrified group. Previous studies demonstrated that enucleation at AI-TI removes significantly less cytoplasm than enucleation at MII; this may be beneficial for the developmental competence of the recipient cytoplast (Lee and Campbell 2006). The ability of an embryo to survive freezing and thawing has previously been reported to be an indicator of embryo quality (Rizos et al., 2001). In the studies reported here, only good-quality oocytes based on morphological criteria were selected, it is possible that selection of the best-quality oocytes here may have influenced cryotolerance, and hence the developmental potential of vitrified/thawed oocytes following SCNT.
Cytogenetic analysis of day 7 blastocysts produced by SCNT revealed a lower proportion of diploid embryos were obtained from vitrified oocytes compared to control; however, the difference was not significant. Published data regarding the cytogenetic status of NT embryos is limited and comparison of results is confounded by a range of factors including technical differences in methodologies between laboratories and the study of different species (Campbell et al., 1999; Li et al., 2004; Slimane-Bureau and King, 2002). The apparent increase in abnormal chromosome constitution reported here may also be attributed to cytoskeletal injuries resulting from vitrification and thawing (Bos-Mickich and Whittingham 1995; Moawad et al., 2010).
In summary, we report for the first time that ovine oocytes vitrified at the GV stage, thawed, and subsequently matured, can be successfully used as cytoplast recipients for SCNT and support a high frequency of development to blastocyst. Although, vitrified GV oocytes developed up to morula stage following parthenogenetic activation with two activation protocols, only the use of strontium produced good-quality blastocysts. The results demonstrate that SCNT and parthenogenetic development form a solid platform to test the developmental potential of vitrified immature ovine oocytes. Further experiments are required to demonstrate the ability of SCNT embryos produced in vitro using vitrified GV oocytes as cytoplasts to establish pregnancy and produce live healthy offspring after transfer to surrogate recipients.
Footnotes
Acknowledgments
Adel R. Moawad was supported by The Ministry of Higher Education, Egyptian Government.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.