
Abstract
Abstract
We investigated the individual and combined effects of leukemia inhibitory factor (LIF) and basic fibroblast growth factor 2 (bFGF2) on the derivation and maintenance of rabbit embryonic stem cell lines isolated from parthenogenetic activated embryos (p-rES). First, we demonstrated that p-rES cell lines can be prevented from differentiation via LIF (STAT3) and bFGF2 (MEK-ERK1/2 and PI3K-AKT) signaling on MEF feeders. High levels of ERK1/2 and AKT activities were crucial for maintaining p-rES cells in an undifferentiated state. Although the p-rES cells under the influence of LIF (500, 1000, and 2000 U/mL) or bFGF2 (5, 10, and 20 ng/mL) alone showed enhanced expression in the pluripotency markers, the highest levels of marker expressions coincided with the simultaneous presence of LIF (1000 U/mL) and bFGF2 (10 ng/mL). The phosphorylation status of LIF and bFGF2 downstream signaling molecules including STAT3, ERK, and AKT was also intensively involved in the maintenance of p-rES cell proliferation and self-renewal. Induced dephosphorylation of STAT3, ERK1/2, and AKT by specific inhibitors caused remarkable losses of self-renewal capacity of p-rES cells. We conclude that bFGF2 and LIF by itself are self-sufficient in maintaining the state of undifferentiation and self-renewal of rabbit p-ES cells, yet are most effective when acting concomitantly.
Introduction
Presently, model animals, such as mice, rabbits, pigs, or nonhuman primates, have become prospective alternatives for the extremely limited human materials for biomedical and clinical studies. Rabbits (Oryctolagus cuniculus) are one of the classical laboratory animals with many advantages over other species. In addition to their short life span and easy to maintain at a large population, they are convenient to breed and to perform surgery due to appropriate body size, and are also the first animal species in which embryo transfer was successfully demonstrated by Walter Heape (Heape, 1890). More importantly, rabbits have been extensively used in research involving human vascular atherosclerosis (Li et al., 2010; Wen et al., 2010), and cardiomyopathy (Jacques et al., 2009; Takato et al., 2010), diabetes (Lin et al., 2007b; Winiarska et al., 2008), and transgenesis for the production of pharmaceutical proteins (Han et al., 2008; Ripplinger et al., 2007).
In hES cells, paracrine factors including bFGFs, BMPs, Wnt, and/or Activin/Nodal are known to be involved in the self-renewal and maintenance of pluripotency (Amit et al., 2000; Dvorak et al., 2005; James et al., 2005; Thomson et al., 1998; Vallier et al., 2005, 2009; Wang et al., 2005; Xu et al., 2005). Undifferentiated hES cells have been reported to express high levels of FGF ligands and their cognate receptors (Brandenberger et al., 2004; Sato et al., 2003; Sperger et al., 2003). It has been generally accepted that LIF is required for maintaining mES self-renewal (Niwa et al., 1998; Smith et al., 1988; Williams et al., 1988), and that STAT3 functions as the key downstream transcription factor in the LIF/gp130 pathway in mES cells (Niwa et al., 1998; Nichols et al., 2001). However, the demand for LIF to maintain ES cell undifferentiation in culture appears to be species-dependent. Previous studies have shown that LIF alone is sufficient to sustain undifferentiation and self-renewal of rES cells on MEF feeders. These rES cells express pluripotency markers and retain the capacity to differentiate into the cell lineages of all three germ layers (Graves and Moreadith, 1993; Chiang et al., 2008; Intawicha et al., 2009). Nevertheless, other paracrine and/or cytokine signalings might also participate in maintaining the stemness and pluripotency of rES cells.
For the fertilized embryo-derived rES cell lines, we have suggested that LIF may have maintained rES cell self-renewal through its conjunctive effects with other paracrine factors secreted by the feeder cells or through the support of other alternative signaling cascades (Intawicha et al., 2009). Other studies also indicated that bFGF is one of the critical factors for the rES cell culture (Honda et al., 2009; Wang et al., 2008). In the present study, we hypothesized that an adequate microenvironment supported by LIF along with bFGF2 and other paracrine factors from the feeders may be advantageous in maintaining the pluripotency and self-renewal of p-rES cells. We demonstrated, for the first time, that LIF and bFGF2 display a collaborative effect on p-rES cell identity when two factors are both present in the culture medium.
Materials and Methods
Reagents and animals
The care and use of animals to recover mature oocytes and fertilized embryos for parthenogenesis and isolation of ES cells complied with the regulation of Institutional Animal Care and Use Committee (IACUC) of National Chung Hsing University, Taiwan, ROC. Chemicals and reagents used were mainly purchased from Sigma-Aldrich Co. (St. Louis, MO, USA) unless otherwise specified. The SCID mice were purchased from BioLasco Taiwan Co., Ltd and raised in accordance with the Guide of the Institutional Animal Use and Care Committee (IACUC) of National Chung Hsing University, Taiwan, ROC.
Establishment and culture of parthenote-derived rES (p-rES) cell lines
Parthenogenetic embryos were derived from New Zealand White rabbit oocytes and activated as described previously (Liu et al., 2002). The activation medium contained 0.3 M D-mannitol (M1902), 0.5 mM MgCl2 (M8266), 0.1 mM CaCl2 (C7902), and 0.1% bovine serum albumin (A8022). First, oocytes were stimulated using three electric pulses at 150 volts/mm for 20 μsec, and incubated with 2.5 mM 6-dimethylaminopurine (6-DMAP, D2629) in M199 medium containing 10% FBS and 1% antimycotics for 30 min. Second, oocytes were electrically pulsed again under the same condition, and then incubated in 6-DMAP for 1 h. Finally, oocytes were transferred to the same medium without 6-DMAP and cultured for 4 days up to the blastocyst stage. The blastocysts were removed of the zona pellucida with 0.5% pronase at 37°C for 3–5 min, washed three times in ES cell medium, and cultured with MEF feeders in the ES cell medium. After removing the trophoblast (TE) cells with a Pasteur pipette, the primary ICM colonies were subcultured and seeded on new MEF feeders (Intawicha et al., 2009).
G-banding for karyotyping p-rES cell lines
Briefly, p-rES cells were incubated with 2 μg/mL of colcemid (Colcemid®, GIBCO, Grand Island, NY, USA) for 6 h at 37°C in 5% CO2, and then were trypsinized, centrifuged, resuspended, incubated in hypotonic solution at 37°C for 6 min, and repeatedly fixed in fixatives (acetic acid:methanol, 1:3) as described previously (Intawicha et al., 2009). After the last fixation, cells were kept in the fixative and dropped onto prechilled glass slides. Chromosome spreads were dried at room temperature (RT) for 2 days and then at 65°C for 4 h, digested with 0.1% trypsin (in 0.8% NaCl) at 37°C for 5 min, stained with 1% Giemsa solution for 20 min prior to microscopic examination, and photographed for analysis. At least 20 distinguishable metaphase spreads with Giemsa-banded karyotypes were selected for analysis.
Detection of alkaline phosphatase (AP) and pluripotency markers
Rabbit ES cells grown on the six-well dishes were rinsed with DPBS prior to fixation in 4% paraformaldehyde (P-6148) solution. After fixation, ES cells were rinsed with DPBS for three times and then stained with AP solution. The stained cells were observed under an inverted microscope after washing with DPBS as described (Intawicha et al., 2009). The expressions of specific protein markers including Oct4, SSEA-4, TRA-1-60, and TRA-1-81, were also detected based on our previous protocols. Finally, epifluorescent dye DAPI (1 μg/mL) was added to DPBST for nuclear staining, followed by two rinses with DPBST before microscopic examination.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNAs of p-rES cell lines (A2 and R3) were extracted using a total RNA extraction kit (Geneaid RT050) and were reverse-transcribed to generate cDNAs for PCR reactions based on the manufacturer's instruction. To each microgram of total RNAs, 2 μL of DNase I, 3 μL of 10 × DNase I buffer, 0.25 μL of RNasin, and 4.75 μL of ddH2O were added and incubated at 37°C for 25 min. After incubation, 1 μL EDTA and oligo-(dT) were added to the tube and reacted at 70°C for 5 min. One microliter of reverse transcriptase (RTase) (Promega M289A, Madison, WI, USA), 10 μL RTase 5 × buffer, 0.25 μL RNasin, 1 μL dNTP, 2.5 μL MgCl2, and 3.25 μL ddH2O were then added to the reaction mixture and incubated at 42°C for another hour. The reaction mixtures were aliquoted and stored at −20°C until the use for PCR reaction. To 25 μL reaction solution containing 2 μL of the template, 18.7 μL of ddH2O were added along with 2.5 μL of 10 × PCR buffer (2.5 mM MgCl2), 0.5 μL of dNTPs (10 mM each; Fermentas R0182, Hanover, MD, USA), 0.3 μL of Taq DNA polymerase (5 U/μL, Geneaid, Taiwan, ROC), 0.5 μL forward primers (10 μM), and 0.5 μL reverse primers (10 μM). The PCR was performed by specific primer sets derived from the aligned conserved regions of the homologous cDNA sequences retrieved from the GenBank database. Primer sequences including sense and antisense, annealing temperatures, and the expected sizes of amplicons are shown in Table 1. Four to 10 μL of amplicons were loaded on a 1.5% agarose gel for separation and visualized under UV light after ethidium bromide staining.
Formation of embryoid bodies (EBs) and induction of teratomas
The p-rES cell colonies were digested with 1 mg/mL dispase (Gibco17105041) for 3 to 5 min at 37°C and split into small clumps by gentle pipetting. They were then cultured in suspension for 4 to 8 days in the medium containing 90% DMEM, 10% fetal bovine serum (FBS), and 2 mM L-glutamine with the medium changed every 3 days. The EBs were continuously cultured in suspension in gelatin-coated six-well dishes for 10 days. The expression of cell markers for the three germ layers was assessed by RT-PCR. For induction of teratomas, nonobese diabetic-severe combined immunodeficiency (NOD-SCID) mice were used as previously described (Intawicha et al., 2009). In brief, 4.1∼20 × 106 morphologically undifferentiated rES cells were subcutaneously injected under the dorsal skin of 7-week-old mice. Five weeks after injection, the teratomas were recovered and fixed in 4% paraformaldehyde for preparation of histological examinations.
Maintenance of p-rES cells in feeder-free culture system
Rabbit p-ES cell lines at passage 17 were treated with 1 mg/mL dispase (Gibco17105041) for 3 to 5 min at 37°C and split into small clumps by gentle pipetting. The cells were maintained on feeder-free and gelatin-coated plates in culture media consisting of 81.5% D-MEM/F12 (Gibco 12400-024), 15% FBS (Gibco10437-028), 4 mM L-glutamine (G8540), 0.5% nonessential amino acids, 0.1 mM β-mercaptoethanol (M7522), and different concentrations (0, 500, 1000, or 2000 U/mL) of murine LIF (mLIF) (Chemicon ESG 1107) or (0, 5, 10, or 20 ng/mL) of human bFGF2 (Prospec CYT-218). The ES cells were analyzed for the expressions of pluripotency markers 4 days after seeding.
Inhibition of LIF and/or bFGF2 signaling pathways in p-rES cells
JAK3 inhibitor I (JAKi, Biosource, PHZ1084) was used to suppress JAK kinase signaling that would otherwise activate STAT3 phosphorylation via LIF stimulation. Inhibitors U0126 and LY294002 (Cell Signaling, 9903 and 9901, Danvers, MA, USA) were used to block MEK-ERK1/2 and PI3K-AKT pathways, respectively. JAK3-inhibitor I (10 μM), 10 μM U0126, 10 μM LY294002, or dimethylsulfoxide (DMSO) were added in medium to culture rES cells for 4 days in order to block the STAT3, ERK1/2, or AKT pathways. After that, the cells were subjected to AP and immunocytochemical staining or Western blotting to detect the expression of pluripotency markers.
Detection of protein expression and phosphorylation
As described previously (Intawicha et al, 2009), p-rES cells were recovered for Oct4 and Nanog expressions by Western blotting. Protein extracts were prepared and stored at −80°C until analysis. Prior to electrophoresis, the extracted protein was boiled and loaded on the 10% SDS polyacrylamide gel. The resolved proteins were transferred onto nitrocellulose membranes (Millipore HAHY00010) for Western blotting. After blotting, the membrane was incubated in blocking buffer (5% chick serum in TBST) for 2 h at RT and then reacted with antibodies against Oct4 (Santa Cruz, SC8628, 1:1000), Nanog (Santa Cruz, SC30331, 1:1000), and β-actin (Cell Signaling, 4967, 1:1000) at 4°C overnight, respectively. The nitrocellulose membrane was washed with 1 × TBST (blocking solution; 200 mM Tris-HCl, 5 M NaCl, 0.05% Tween-20, pH 7.4) and then subjected to the treatment of HRP-conjugated secondary antibodies (Anaspec, 28169) for 1 h for visualization with a Super-Signal West chemiluminescent substrate kit (Thermo Scientific, West Palm Beach, FL, USA). The intensity of protein signals in three duplicates was determined by using the Image J software and by Java software (Version 1.42 for Windows). Beta-actin serves as a loading control to normalize the expressions of Oct4 and Nanog.
To detect the phosphorylation statuses of STAT3, ERK1/2, and AKT, p-rES cells were harvested in 0.05% trypsin-EDTA (1 mM) for protein extraction and then electrophoresis as described previously (Intawicha et al., 2009). For Western-blot analysis, anti-STAT3 (Cell Signaling, 9132, 1:1000), anti-phospho-STAT3 (Serine 727; Cell Signaling, 9134, 1:1000), anti-ERK1 (Santa Cruz, SC94, 1:1000), anti-phospho-MEKK/ERK1/2 (Cell Signaling, 9101, 1:1000), anti-AKT (Cell Signaling, 9272, 1:1000), anti-phospho-AKT (Ser 473; Cell Signaling, 1:1000), and anti-β-actin (Cell Signaling, 4967, 1:1000) antibodies were used. Similarly, primary antibodies were incubated overnight with the blot and then reacted with HRP-conjugated secondary antibodies (KLP, 474-1516) except that anti-phospho-AKT antibody was reacted with HRP-conjugated secondary antibodies (Cell Signaling, 7067) for 1 h. Protein signals on the blots were visualized and analyzed as described above.
Statistical analysis
Data in percentages were normalized by arcsine transformation prior to statistical analysis by using the least significant difference (LSD) or chi-square test. The results are presented as means ± standard error of the mean (SEM). Statistical significance was considered when p < 0.05.
Results
Derivation of rES cell lines from fertilized and parthenogenetic embryos
The efficiencies of deriving ES cell lines differed between using fertilized embryos and parthenotes (fertilized embryos vs. parthenogenetic embryos) as shown in Table 2. A total of 64 blastocysts from fertilized embryos and 76 from parthenotes were seeded onto MEF feeders. Rabbit ES cell clones successfully established from fertilized embryos outnumbered those from parthenotes (11 vs. 3) (p < 0.05). The primary colonies derived from parthenotes were successfully grown in culture as shown in Figure 1A. After removal of the TE cells covering the ICM clump, the produced ES cell colonies can be further passaged. Three out of the 20 ICM colonies were continuously cultured into p-rES cell lines with morphology common to f-rES cells. The p-rES cell lines expressed AP, Oct4, SSEA-4, TRA-1-60, and TRA-1-81 as examined by immunocytochemical staining. On the other hand, by RT-PCR analysis, both p-rES (A2 as representative) and f-rES (R3 as representative) lines expressed pluripotency genes Oct4, Nanog, and Sox2 (Fig. 1B). Cytogenetic analyses of the p-rES cells (A2, passage 14) indicated that 66.7% of the cells examined were of normal karyotype (2n = 44).

Characterization of parthenote-derived rabbit embryonic stem (p-rES) cell lines isolated from parthenogenetically activated embryos of New Zealand White rabbits. The p-rES cell line (A2) stained positively for alkaline phosphatase (AP), and expressed Oct4, SSEA-4, and the keratan sulfate antigens TRA-1-60 and TRA-1-81 as detected by immunocytochemistry (
Chi-square tests were performed. Significant differences were detected between treatment groups (p < 0.05).
Five replicates.
Within the column, numbers with different superscripts differed (p < 0.05).
Expressions of imprinted genes and differentiation in EBs and teratomas
Examination of the transcripts of imprinted genes revealed that while f-rES cells (R3) expressed H19, Snrpn, and Igf2 genes, p-rES cell lines (A2) expressed only the maternally imprinted H19 (Fig. 1C).
In the presence of serum, p-rES cells also formed EBs in suspension culture (Fig. 1D). Our RT-PCR results showed that EBs derived from four p-rES cell lines, similar to f-rES cells, expressed marker genes of the three germ layers (Fig. 1E), that is, Desmin (mesoderm), Gata4 (endoderm), and Map2 (ectoderm). Nonobese diabetic (NOD)-SCID mice injected with the p-rES cells line (A2) formed teratomas 5 weeks after injection (Fig. 2A and B). Histological examination showed that the teratoma contained a variety of tissue cell types including salivary glands (ectoderm, Fig. 2D), skin (ectoderm, Fig. 2E), respiratory tube (endoderm, Fig. 2F), cartilage (mesoderm, Fig. 2G), and smooth muscle (mesoderm, Fig. 2H).
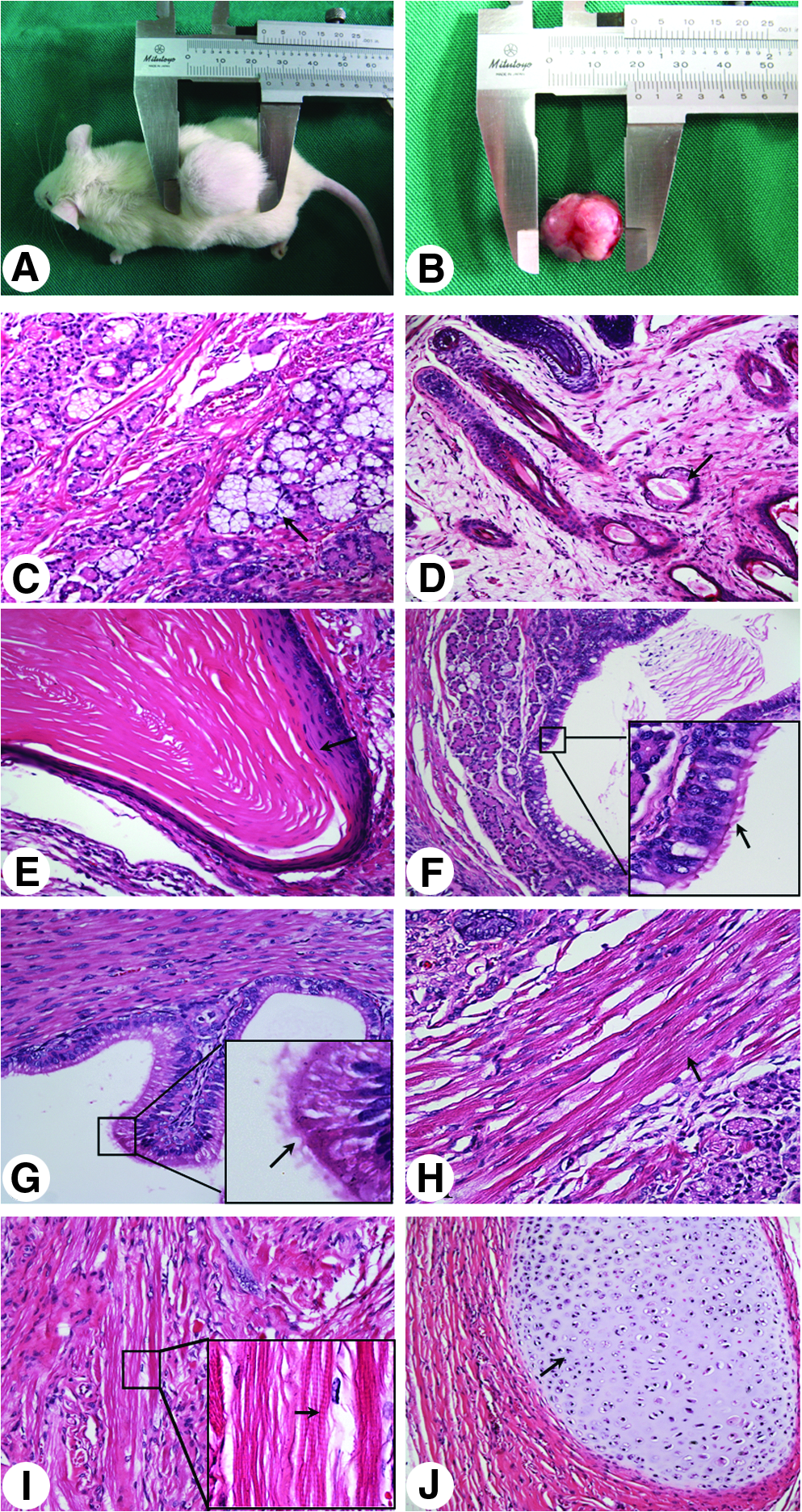
Teratoma formation of the parthenote-derived rabbit embryonic stem (p-rES) cells 5 weeks after subcutaneous injection into NOD-SCID mice. (
Basic FGF2 and/or LIF pathways sustains self-renewal of p-rES cells grown on MEF feeders
In this study, we examined the roles of ERK1/2, AKT, and the downstream regulators of the FGF pathway in sustaining the self-renewal of two representative p-rES cell lines (A2 and A4). Western blot analyses confirmed that expressions of the ERK1/2 and AKT upon bFGF2 stimulation worked as the key downstream mediators in p-rES cell lines. Phosphorylations of ERK1/2 and AKT were also observed in undifferentiated p-rES cell lines under bFGF2 stimulation (20 ng/mL) for either 10 or 30 min (Fig. 3A).

LIF and bFGF2 activate MEK/ERK and PI3K/AKT signaling pathways that maintain the pluripotency of p-rES cells. (
In the presence of LIF alone, p-rES cells showed similar levels of STAT3, AKT and ERK1/2 phosphorylation with the untreated cells. On the contrary, with bFGF2 in the medium, p-rES cells showed somewhat greater levels of ERK1/2 (A4) and AKT (A2) phosphorylation than those cultured in the medium lacking FGF2 and LIF (Fig. 3B and C). Interestingly, p-rES cells cultured in the presence of both LIF and bFGF2 showed significantly increased levels of STAT3, ERK1/2, AKT phosphorylation, and in turn, better cell propagation than the untreated p-rES cells (p < 0.05).
LIF or bFGF2 alone maintained p-rES cell self-renewal under feeder-free condition
To eliminate the effects of paracrine factors secreted by the feeder cells on p-rES cell survival and propagation, the feeder-free culture system was adopted to reexamine the controversial LIF-dependency. Two p-rES cell lines (A2 and A4) at passage 18 were treated with 500, 1000, and 2000 U/mL of LIF, respectively. Those p-rES cells with null LIF treatment lost their defined colony morphology at day 4 of culture, whereas the LIF-treated groups sustained normal colonies. Expression levels of the pluripotency markers, including AP, TRA-1-60, and TRA-1-81 rose in proportion to the amount of LIF supplemented in p-rES cell A2 line (Fig. 4A). In both cell lines (A2 and A4) examined, Oct4 and Nanog expressions went up when LIF was supplemented, most notably at 2000 U/mL (Fig. 4B and C).
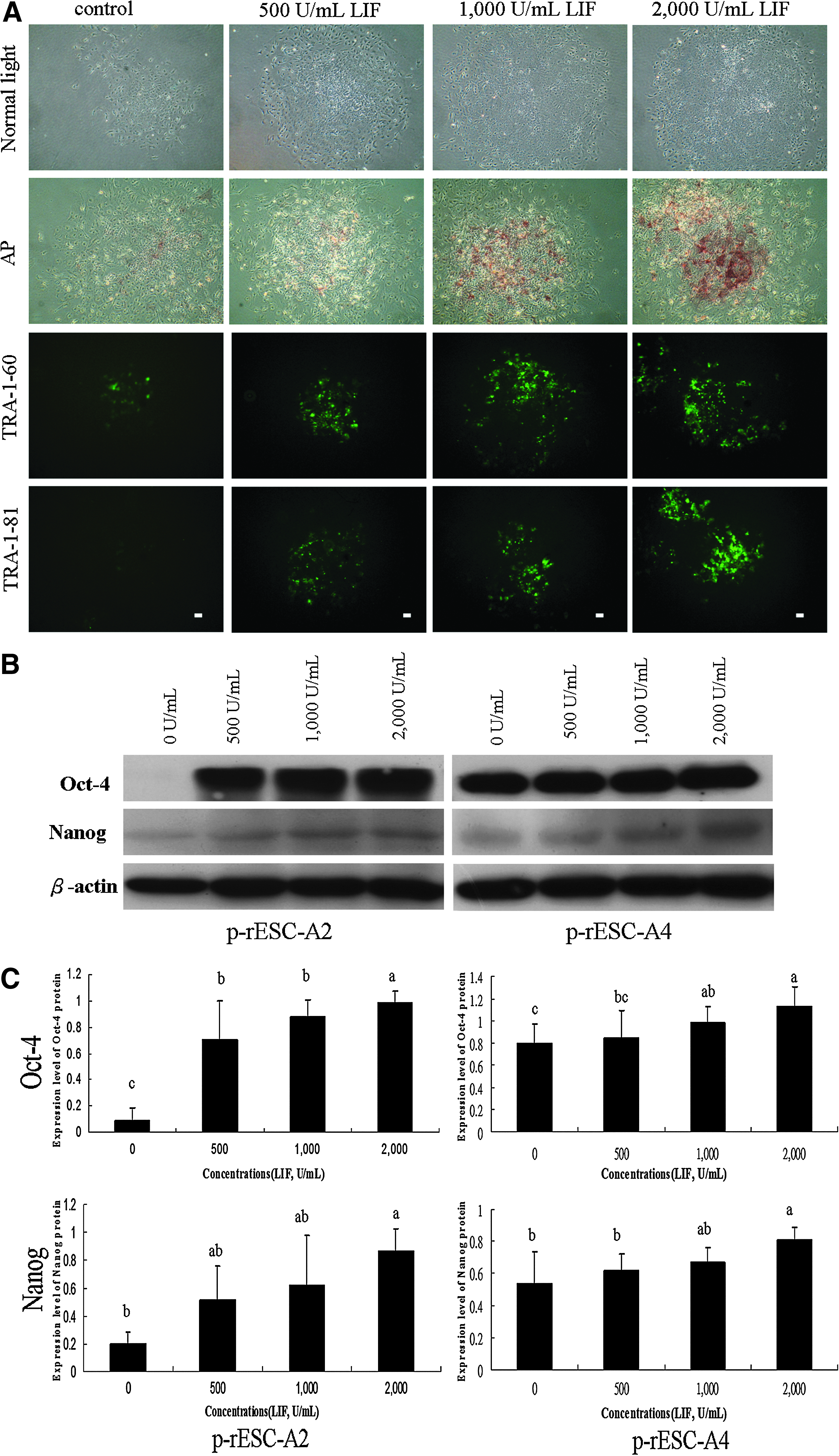
Leukemia inhibitory factor (LIF) maintains the pluripotency of parthenogenetic embryo-derived rabbit ES (p-rES) cells in the feeder-free culture system. Representative images show the colony morphology of p-rES cells (A2, passage 18) cultured in the FBS-containing medium supplemented with (500, 1000, and 2000 U/mL) or without LIF for 4 days in the feeder-free condition. (
Similarly, when the feeder-free p-rES cells were supplemented with bFGF2 (5, 10, and 20 ng/mL), they all remained compact and retained better morphologies than those untreated control (Fig. 5A). Moreover, p-rES cells showed higher levels of TRA-1-60, TRA-1-81, and AP activity in the bFGF2-treated groups (Fig. 5A). In the presence of 20 ng/mL bFGF2, p-rES cells showed significantly higher levels of Oct4 and Nanog than those control cells (p < 0.05) as evident by Western blot analyses (Fig. 5B and C).

Basic fibroblast growth factor 2 (bFGF2) maintains the pluripotency of parthenogenetic embryo-derived ES (p-rES) cells in the feeder-free culture system. Representative images of the colony morphology of p-rES cell line (A2, passage 18) cultured in the FBS-containing medium supplemented with (5, 10, and 20 ng/mL) or without bFGF2 for 5 days in the feeder-free condition. The expressions of AP, TRA-1-60, and TRA-1-81 markers were all detected in the undifferentiated colonies (
Effects of the combined LIF and bFGF2 treatment on p-rES cell self-renewal
Two p-rES cell lines were allocated and cultured in one of the four treatment groups: (1) control, without LIF and bFGF2; (2) addition of 1000 U/mL LIF, (3) addition of 10 ng/mL bFGF, and (4) addition of 1000 U/mL LIF and 10 ng/mL of bFGF2, in a feeder-free culture system. As presented previously, we found that p-rES cells when treated with LIF or bFGF2 alone only maintained normal colony morphology in both cell lines for 2–3 days. On the other hand, cells simultaneously treated with LIF and bFGF2 exhibited better compact colony morphology than those individually treated with LIF or bFGF2 (Fig. 6A). In addition, cells treated with LIF and bFGF2 together obtained marked increase in the expressions of AP, TRA-1-60, and TRA-1-81. In contrast, AP, TRA-1-60, and/or TRA-1-81-positive colonies were hardly detectable when both LIF and bFGF2 were absent in the medium (Fig. 6A). Similar expression patterns were also observed for the Oct4 and Nanog protein expressions (p < 0.05) as revealed by Western blot analyses (Fig. 6B and C).
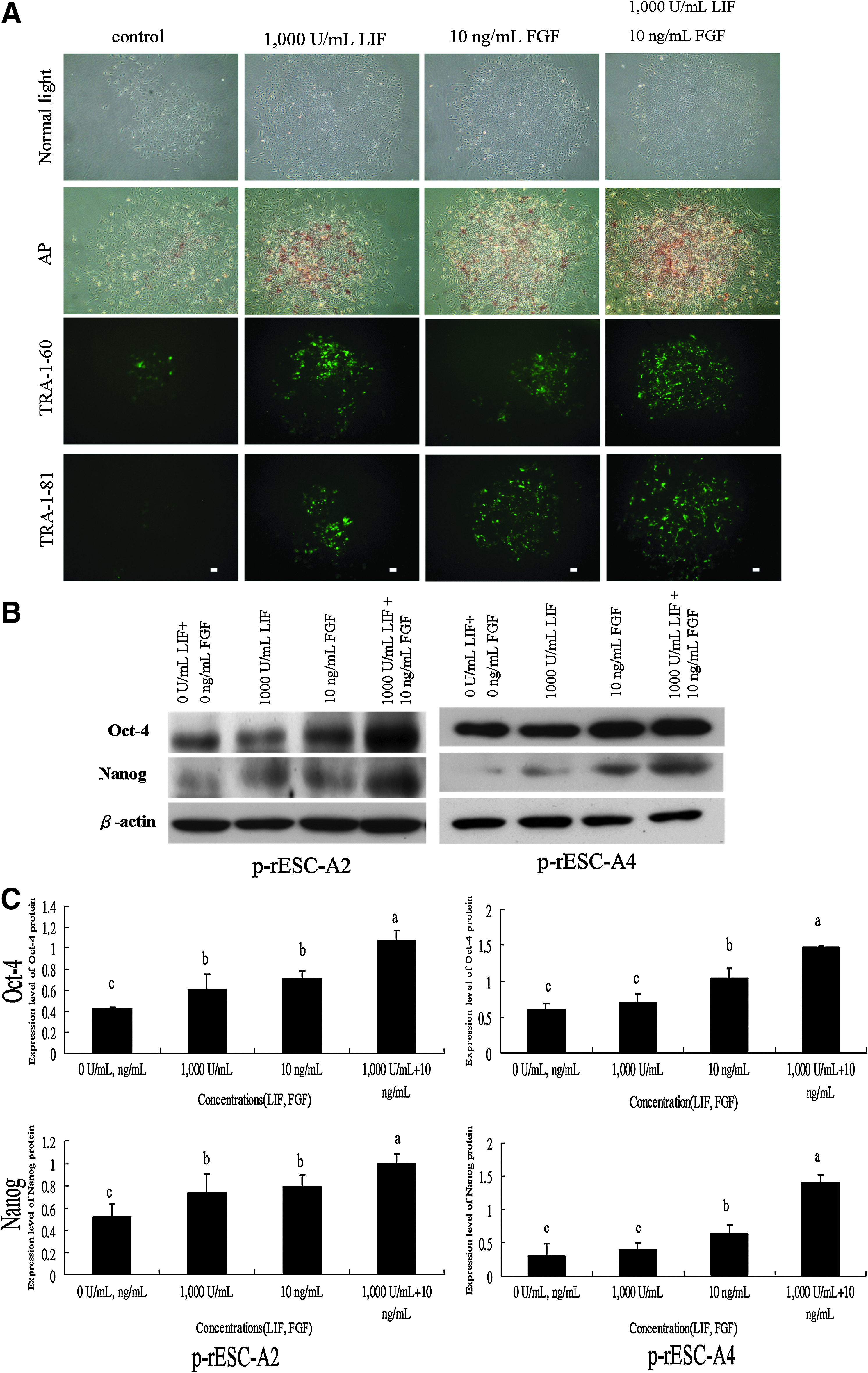
The effect of LIF and bFGF2 combined treatment on the pluripotency marker expressions of p-rES cells (A2, passage 18) in the feeder-free system. The p-rES cells were cultured in the FBS-containing medium in the presence of LIF and/or bFGF2 for 4 days. The expressions of AP activity and TRA-1-60/81 were detected with a much stronger level in the combined treatment group (
Inhibitory dephosphorylation of STAT3, MEK/ERK, and AKT blocked self-renewal in p-rES cells
Inhibitory drugs against LIF and FGF downstream signaling were employed to suppress LIF and FGF stimulations. Inhibitors for JAK kinase (JAKi), MEK/ERK1/2 1/2 (U0126), or PI3K/AKT (LY294002) were used to treat p-rES cells grown in the feeder-free culture supplemented with LIF (1000 U/mL) and bFGF2 (10 ng/mL). The p-rES cell colonies failed to maintain normal morphology if either inhibitor was present in the medium. There was also a tendency that coexistence of multiple inhibitors exacerbated the growth of p-rES cells. Protein expression levels of the pluripotency markers, Oct4 and Nanog, were also reduced, most notably when all three inhibitors were present in the same culture (Fig. 7A–C).

The specific kinase inhibitors in the LIF and bFGF2 pathways suppress the expressions of pluripotency markers in parthenogenetic embryo-derived rabbit ES (p-rES) cells. The p-rES cell line (A2, passage 18) were cultured in ES cell medium supplemented with LIF and bFGF2. The six treatments labeled by Arabic numbers 1–6 throughout the panels were shown only on top of
Discussion
Parthenogenetically activated embryos derived by artificial stimuli of oocytes without undergoing fertilization event possess genetic materials of entirely maternal origin. It has been generally accepted that a PA embryo fails to develop to term due to a lack of paternal genome or normal genomic imprinting (Fundele et al., 1990; Kono et al., 2004). However, parthenote-derived ES cells with the advantage of less ethical controversies had become a proper substitute for those derived from somatic cell-cloned embryos (Kaufman et al., 1983; Kim et al., 2007b; Sánchez-Pernaute et al., 2005; Vrana et al., 2003). In addition, autologous stem cells and/or parthenote-derived ES cells have been proved to be less prone to immunological rejection after transplantation (Gong et al., 2008). Recently, hES cell lines have been successfully derived from the parthenogenetic embryos (Brevini et al., 2009; Kim et al., 2007b; Lin et al., 2007a; Mai et al., 2007; Revazova et al., 2007, 2008). The establishment and use of p-ES cells may accelerate the development of personalized medicine. In the present study, three p-rES cell were successfully derived from parthenogenetic embryos. Similar to f-rES cells, these p-rES cells displayed the typical f-rES cell morphology and the same expression profiles of the surface antigens including SSEA-4, TRA-1-60, and TRA-1-80 (Fig. 1A) in addition to the pluripotency markers Oct4, Sox2, and Nanog (Fig. 1B). These p-rES cells are also capable of forming teratomas in NOD-SCID mice after subcutaneous implantation, in which tissues with the progenitors of three germlayers were disclosed through systemic histological examinations (Fig. 2). The results are similar to our findings in f-rES cells (Intawhicha et al., 2009). It validated that p-rES cells could differentiate into cell lineages of three germ layers after proper induction.
Rabbit ES cells derived from PA embryos can be maintained under a similar culture condition to hES cells with low FGF concentration (4 ng/mL) as reported by Thomson et al. (1998). Previous reports also indicated that MEF feeder culturing condition with a supplementation of 4–8 ng/mL of bFGF2 (Fang et al., 2006) or 1000 U/mL LIF (Intawicha et al., 2009) was sufficient to derive and maintain rES cells. In the present study, we directly examined FGF signaling associated with the mitogen-activated protein kinases ERK1/2. We found that the MEK/ERK signaling was essential for maintaining the pluripotency and self-renewal of p-rES cells, similar to hES cells (Armstrong et al., 2006; Gong et al., 2008) and rES cells (Intawicha et al., unpublished data) on MEF feeders.
It has been well known that both MEK/ERK1/2 and PI3K/AKT signaling pathways are important in modulating cell death or survival in human somatic cell (Ballif and Blenis, 2001). For ES cells, inhibition of PI3K/AKT signaling augments LIF-induced phosphorylation of ERKs and induces differentiation, whereas simultaneous inhibition of PI3K/AKT and MEK/ERK1/2 reverses the negative effect of PI3K inhibition on mES cell self-renewal, suggesting that the elevated ERK1/2 activity observed upon PI3K inhibition leads to differentiation (Paling et al., 2004). In our results, significant activation of MEK/ERK1/2 and PI3K/AKT signaling by bFGF2 and/or LIF was observed (Fig. 3B and C). Therefore, it has become clear that ERK1/2 and AKT signalings play an important role in maintaining p-rES cells in a state of undifferentiation. Noteworthy is that Honda et al. (2009) demonstrated that p-AKT was independent of bFGF2 dosages in their f-rES cells, which was contradictory to our current finding. The reasons behind the disagreement could be multifactorial; first of all, the animal subspecies used were different: the Japanese White rabbit in the study of Honda's group versus the New Zealand white rabbit in ours. Second, they used feeder cells at the confluency of 6 × 103 cells/cm2 as opposed to our 2 × 104 cells/cm2. Finally, the experimental designs were different; in this study the p-rES cells were subjected to serum starvation for 12 h to eliminate the effects of other confounding paracrine molecules. We then used DMEM/F-12 supplemented with LIF or bFGF2 (no FBS) to activate rES cells. On the contrary, in their study the p-AKT was measured under normal culture condition in ES medium supplemented with bFGF2, without undergoing starvation.
As in previous studies, LIF signaling pathway maintains the symmetrical self-renewal in mES cells (Smith et al., 1988) but not in hES cells (Thomson et al., 1998). Our previous studies also showed that LIF signaling was sufficient to maintain self-renewal of f-rES cells, and the major players in the LIF signaling, including LIFR/gp130, JAK kinases, and STAT3, were all expressed in f-rES cells. Such LIF-dependent characteristic of rES cells is similar to that of mES cells (Intawicha et al., 2009). In this study, we have successfully maintained p-rES cell lines, using the medium supplemented with bFGF2 or LIF alone. These cell lines expressed all the pluripotency genes or protein markers as examined in hES cells (Amit et al., 2000; Thomson et al., 1998) and remained unspecialized during long-term culture. It appears that both signaling pathways are also effective in p-rES cells in culture.
To eliminate the effects of other confounding paracrine molecules in culture, we tested the feeder-free system to highlight LIF and FGF signaling in p-rES cells. First, effects of different concentrations of LIF on the maintenance of rES cells were evaluated. We found that upon shortage of LIF rES cells started to differentiate and had difficulties in sustaining self-renewal of colonies as determined by their morphology and by the expressions of AP or TRA-1-60 and TRA-1-81, a condition that can be prevented by adding 500 U/mL or more of LIF (Fig. 4A). Expressions of Oct4 and Nanog proteins apparently increased with the amount of LIF added in culture (500, 1000, and 2000 U/mL) (p < 0.05) (Fig. 4B and C). This dosage-dependent feature also existed in the culture when bFGF2 was supplemented alone (Fig. 5). Most strikingly, in the presence of both LIF and bFGF2, p-rES cell colonies maintained a normal morphology and exhibited apparently stronger AP activity and TRA-1-60/TRA-1-81 expressions in comparison with those cells solely supplemented with LIF or bFGF2 in culture (Fig. 6). We also found that the phosphorylation of LIF and bFGF2 downstream molecules, including STAT3, ERK1/2, and AKT, was drastically influenced by the presence of LIF or/and FGF. Expression levels of pluripotency markers and signaling proteins were also affected by the presence of various inhibitors in the feeder-free culture condition (Fig. 7). These results suggest that LIF and bFGF2 both act to maintain cell pluripotency and self-renewal in rES cells, even though more complicate cascades involving additional signaling molecules could not be completely excluded. It is worth mentioning that all the p-rES cell pluripotent characteristics could be maintained by a single factor (LIF or bFGF2) at a rather basal level, and reinforced to some extent by the coexistence of both factors or by a single factor at high concentrations.
To summarize our current study, upon binding of LIF to its heterodimer receptor (LIFR) and gp130, JAK/STAT3 become activated to phosphorylate and dimerize STAT3 for sustaining undifferentiation of p-rES cells as in mES cells. On the other hand, our results also demonstrated that bFGF2 functioned to activate MEK/ERK1/2 and PI3K/AKT signaling pathways, which was as significant to hES cells as to p-rES cells in self-renewing. When LIF and FGF pathways were both activated, the levels of MEK/ERK1/2 and PI3K/AKT phosphorylation increased to the largest extent, perhaps, due to the alleged phosphorylation of ERK1/2 via Grb2 (growth-factor-receptor-bound protein 2) and AKT/PI3K exerted by LIF in mES cells (Burdon et al., 2002; Niwa et al., 2009).
A working model to describe the joint effects of LIF and bFGF2 signaling to maintain rabbit ES cell proliferation in culture is proposed as depicted in Figure 8. When LIF and FGF are both supplemented, the phosphorylation of MEK/ERK1/2 and PI3K/AKT increases to the largest extent, possibly due to the fact that LIF also has an effect on the phosphorylation of ERK1/2 via Grb2 (growth-factor-receptor-bound protein 2). This activation indicated by a dashed arrow pointing to Ras may be dependent on the phosphorylation of the SH2-domain-containing cytoplasmic tyrosine phosphatase SHP2 and awaits further studies (Annerén, 2008; Burdon et al., 2002). The LIF signaling also interlinks with the FGF2 signaling via PI3K/Akt pathway, which has been shown to be important for the proliferation, survival, and self-renewal of mES cells (Paling et al., 2004; Watanabe et al., 2006). This mutual concoction between LIF and FGF signaling may explain why the additions of both LIF and bFGF showed weak or inconsistent synergistic effects on the cell pluripotency and self-renewal in p-rES cells (Figs. 3C and 6C).
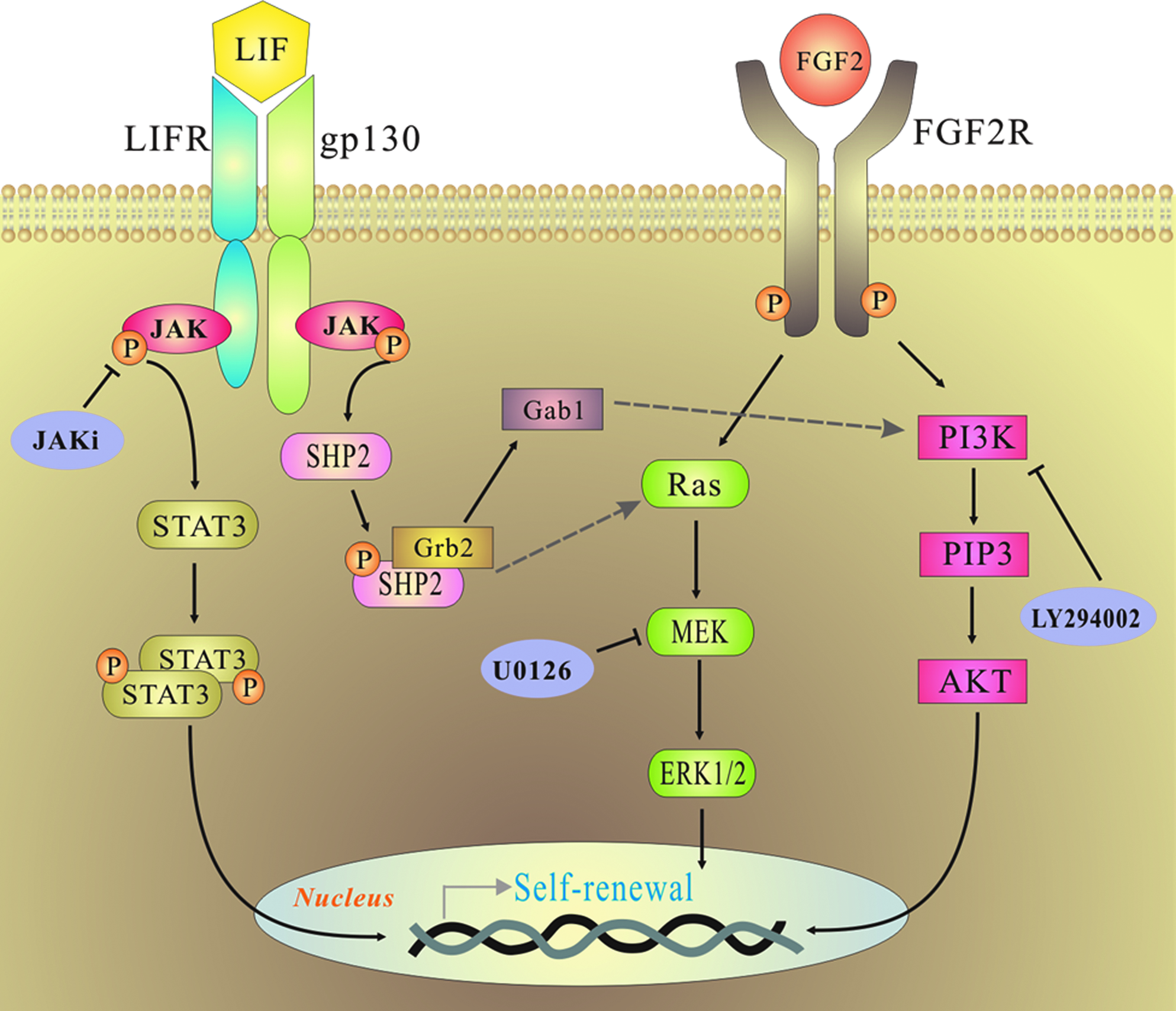
The proposed model illustrates the collaborative integration of LIF and bFGF2 signaling pathways to sustain stemness of rabbit ES cells. The arrow-ended lines indicate the potential route for passing down the positive stimulations from their upstream molecules, and the hammer-ended lines are the positions at which the inhibitors are effective. Arrows with dashed line indicate some potential routes awaiting further confirmation. (See color version of this figure at www.liebertonline.com).
It is not unexpected that different signaling molecules, such as BMP, Wnt, Activin/Nodal, and/or other TGF family ligands (Heape, 1890; Honda et al., 2009; Wang et al., 2008) secreted by the feeder cells, may also partake in maintaining rabbit ES cell pluripotency, self-renewal, and proliferation. Nevertheless, we conclude that LIF or bFGF2 alone is sufficient to establish and maintain parthenote-derived rabbit ES cell lines. When both factors are supplemented in culture, an additive effect in sustaining the stemness of rabbit ES cell lines can be achieved.
Footnotes
Acknowledgments
This study was supported by a grant from National Science Council, Executive Yuan (NSC# 97-2313-B-005-003-MY3), Taiwan, ROC.
Author Disclosure Statement
The authors declare that no conflict of financial interest exists.