
Abstract
Abstract
We report here the establishment and characterization of putative porcine embryonic stem cell (ESC) lines derived from somatic cell nuclear transfer embryos (NT-ESCs). These cells had a similar morphology to that described previously by us for ESCs derived from in vitro produced embryos, namely, a polygonal shape, a relatively small (10–15 μm) diameter, a small cytoplasmic/nuclear ratio, a single nucleus with multiple nucleoli and multiple lipid inclusions in the cytoplasm. NT-ESCs could be passaged at least 15 times and vitrified repeatedly without changes in their morphology, karyotype, or Oct-4 and Nanog expression. These cells formed embryoid bodies and could be directed to differentiate in vitro to cell types representative of all three germ layers. Following their injection into blastocysts, these cells preferentially localized in the inner cell mass. In conclusion, we have isolated putative porcine ESCs from cloned embryos that have the potential to be used for a variety of applications including as a model for human therapeutic cloning.
Introduction
To this end we have already developed a relatively efficient cloning method for pigs and have used this to produce cloned pigs using unmodified and genetically modified fetal and adult fibroblasts (Beebe et al., 2007a; Boquest et al., 2002; Harrison et al., 2004; Nottle et al., 2007). Furthermore, we have recently reported that the efficiency of this method can be improved by treating cloned embryos with the histone deacetylase inhibitor Trichostatin A (TSA; Beebe et al., 2009b). TSA has also been shown to increase the efficiency with which ESCs can be isolated from cloned embryos in mice (Kishigami et al., 2006). The aim of the present study was to use these technologies to determine whether ESCs can be isolated from cloned porcine embryos treated with TSA.
Materials and Methods
Maturation of pig oocytes in vitro
Porcine ovaries from slaughtered mature sows were collected from a local abattoir and transported to the laboratory in phosphate-buffered saline (PBS) solution, containing 1% of an antibiotic–antimycotic solution (Invitrogen, New Zealand) at a temperature between 33 and 37°C. Follicles with a diameter between 3 and 6 mm were aspirated with a 21-G needle, through which a constant suction (1 L/min) was applied, and the follicular contents pooled in a collection tube. Cumulus–oocyte complexes (COC) with at least three uniform layers of compact cumulus cells were recovered from the collected fluid and matured for 40–42 h in groups of 50 in 500 μL of M199 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 5 ng/mL insulin, 0.5 mM cysteamine, 0.2 mM Na-pyruvate, 75 mg/mL Penicillin-G, 50 mg/mL Streptomycin sulphate, 10 ng/mL hEGF (all from Sigma, St. Louis, MO, USA), 5 mg/mL Follicle Stimulating Hormone (ICP Bio, New Zealand), and 10% sow follicular fluid under mineral oil (Sigma) in a humidified atmosphere of 5% CO2, 5% O2, and 90% N2 at 38.5°C. Sow follicular fluid was collected from follicles with diameter between 3 and 6 mm, stored at −70°C, thawed, and filter sterilized (Millipore, Badford, MA, USA; filter pore size 0.22μm) immediately prior to use.
Somatic cell nuclear transfer procedure
The oocyte enucleation and reconstruction protocol used was as described previously (Beebe et al., 2009b). Briefly, enucleated oocytes were produced by removing the cumulus cells from in vitro matured COCs by manual pipetting. Oocytes were stained with Hoechst 33342 (7.5 μg/mL; Sigma) and the first polar body and adjoining cytoplasm, presumptively containing the metaphase plate removed. Successful enucleation was confirmed by brief exposure to UV light to check for the removal of the polar body. A single adult skin fibroblast of Duroc boar D211 cell line was placed in the perivitelline space of each enucleated oocyte. Couplets were then fused with one DC pulse of 220 V/mm for 60 μsec. Approximately 1.5 h later, couplets were activated with two DC pulses of 150 V/mm for 60 μsec given 1 sec apart. Reconstructed embryos were then cultured with 7.5 mg/mL cytochalasin B for 3 h in modified NCSU23 (Beebe et al., 2007b) containing 0.2 mM pyruvate, 5.7 mM lactic acid, 0.6 mM glucose (all from Sigma) and MEM-nonessential amino acids (Invitrogen). Embryos were then cultured in this media for the first 3 days followed by NCSU23 medium containing 5.6 mM glucose (Sigma) and MEM nonessential and essential amino acids (Invitrogen) to day 6. Culture was conducted in 50 μL droplets under mineral oil (Sigma) in a humidified atmosphere of 5% CO2, 5% O2, and 90% N2. A low oxygen tension was chosen because we have previously shown that this is superior compared with atmospheric oxygen for the isolation and culture of porcine fibroblasts (Harrison et al., 2004) and embryo development (Booth et al., 2005).
Experimental design
Experiment 1 examined the effect of TSA on the development of NT embryos and primary outgrowth formation compared with controls. In the experimental group NT embryos were treated with 50 nM TSA for 24 h right after activation (3 h in the presence of cytochalasin B and 21 h without cytochalasin B) and then cultured as described above until day 7. Control NT embryos were not treated with TSA after activation and were cultured as described above. NT embryos from control and TSA-treated groups were then plated onto mitotically inactivated mouse embryonic fibroblast (MEF) to determine the effect of TSA treatment on the establishment of homogeneous primary embryonal outgrowths. These were assessed morphologically and examined for Oct-4 and Nanog coexpression using immunofluorescence.
In Experiment 2, primary embryonal outgrowths isolated from control and TSA treated NT day 7 embryos were used to derive NT-ESC lines. NT-ESC lines were considered to be established if they survived repeated passaging and vitrification at least once. These lines were characterized in terms of Oct-4 and Nanog expression, their ability to form embryoid bodies and undergo directed differentiation to cells representative all three germ layers.
Establishment of embryonal outgrowths and isolation putative porcine NT-ESC lines
Primary embryonal outgrowths were established from control and TSA-treated day 7 in vitro cultured NT embryos. The zona pellucida was removed on day 5 with Tyrode's solution (Sigma) in order to prevent any damage to the cells as a result of the blastocyst prematurely extruding out through the slit in zona pellucida made during enucleation. NT embryos with prominent inner cell masses (ICMs) were placed on mitotically inactivated MEF feeder layers and gently pressed onto the feeder layer using a 30-G needle. The isolation media used consisted of αMEM medium with ribo- and deoxyribonucleosides supplemented with 10% Serum Replacement (SR), 20 ng/mL bFGF, 20 ng/mL human recombinant EGF, 1× Insulin–Transferrin–Selenium solution, 55 μM 2-Mercaptoethanol, 1× MEM nonessential amino acids, 1× Glutamax (all from Invitrogen) 10 ng/mL human recombinant LIF (Millipore), and 10 ng/mL Activin A (R&D Systems, Minneapolis, IN, USA). Cultures were conducted in a humidified atmosphere of 5% CO2, 5% O2, and 90% N2 at 38.5°C. The media were changed every 2–3 days. After 12–15 days of culture primary outgrowths that had reached 2–5 mm in diameter were mechanically passaged onto fresh feeder layers. This was done by cutting outgrowths into approximately 80–100 micron square pieces using a hand-drawn glass Pasteur pipette, transferring these onto new feeder layers, and then gently pressing the pieces into the feeder using a 30-G needle. Colonies were passaged every 10–12 days. Putative NT-ESC cells in primary outgrowths were identified according to morphological criteria described previously for bovine ESCs (Cibelli et al., 1998; Mitalipova et al., 2001) and for putative porcine ESCs (Vassiliev et al., 2010a, 2010b), namely, polygonal in shape, a relatively small (10–15 μm) diameter, a small cytoplasmic/nuclear ratio, a single nucleus with multiple nucleoli, and multiple lipid inclusions in the cytoplasm. Homogeneity of outgrowths was assessed morphologically initially and then examined with immunofluorescence analysis for Oct-4 and Nanog expression. Expression of Gata-6 and Cdx-2 as markers of extraembryonic endoderm and trophoblast cells, respectively, were also examined using immunofluoresence. Cell lines were vitrified at passage 2 and then thawed. Cell lines that survived vitrification and warming and continued to grow were considered viable.
Vitrification and warming of porcine ESC lines
NT-ESC colonies were dissected into smaller pieces as described above. Pieces were vitrified using solid surface vitrification using the Cryologic Vitrification System (CVM; Cryologic Pty. Ltd., Victoria, Australia). The solutions used to vitrify and warm the embryos were warmed to 39°C prior to use. ESC colony pieces were held in 1 mL of the base medium composed of αMEM medium, 2.5% vol/vol HEPES buffer solution (both from Invitrogen), and 20% fetal bovine serum (FBS). Between 6 and 10 pieces were then washed in fresh base medium, incubated for 1 min in 1 mL of the base medium containing 10% (vol/vol) ethylene glycol (Sigma) and 10% (vol/vol) dimethyl sulphoxide (Sigma), and then washed for about 1 min in 1 mL of the vitrification medium composed of base medium, 0.4 M sucrose, 20% ethylene glycol, and 20% dimethyl sulphoxide. The ESC colony pieces were then loaded into a 3-μL droplet of vitrification medium on a nylon hook and the droplet quickly touched onto the surface of a metal block that had been cooled by partial immersion in liquid nitrogen. The nylon hooks were then covered with cooled plastic sleeves, plunged into liquid nitrogen, and stored. The ES cell colony pieces were warmed by stirring the vitrified droplet containing the colony pieces in approximately 1 mL of the base medium containing 0.2 M sucrose and holding them in that drop for approximately 1 min. They were then incubated in 1 mL of the base medium containing 0.1 M sucrose for 5 min, followed by the base medium only for 5 min, and then a fresh 1 mL of the base medium for a further 5 min. The warmed pieces were plated onto fresh feeder layer.
Karyotype analysis
NT-ESC colonies were treated with 0.05 μg/mL colcemid (Invitrogen) for 24 h at 37°C to arrest the cell cycle. Colonies were picked from the feeder layer and dissociated with TrypLE Express solution (Invitrogen), treated with hypotonic solution (0.075 M KCl; Invitrogen), and then fixed in methanol–acetic acid (3:1). Cells were then sent to the Cytogenetics, Genetics and Molecular Pathology Laboratory at Women's and Children's Hospital (Adelaide, Australia) for karyotype analysis.
Immunofluorescence analysis of expression of pluripotent markers and extraembryonic cell markers in primary outgrowths and NT-ESC lines
Primary embryonal outgrowths and NT-ESC colonies were fixed with 4% PFA and permeabilized with 0.3% saponin (Merk,, Germany) and analyzed for expression of Oct-4, Nanog, Gata-6, and Cdx-2. The primary antibodies were rabbit antibody to Nanog (1:300 dilution; Millipore), goat antibody to Oct-4 (1:100 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), rabbit antibodies to Gata-6 (1:100 dilution; Santa Cruz), and rabbit antibodies to Cdx-2 (2 μg/mL; Millipore). After washing, primary outgrowths or NT-ESCs were incubated for 30 min with secondary antibodies at room temperature. Secondary antibodies were chicken antirabbit antibody conjugated to Alexa-647 (1:500 dilution; Invitrogen), donkey antigoat antibody conjugated to Alexa-488 (1:1000; Invitrogen) and donkey antirabbit antibodies conjugated to Alexa-546 (1:1000; Invitrogen). Imaging analysis was done using a confocal laser microscope SP5 (Leica, Allendale, NJ, USA) using Z-stack sectioning with 0.4-μm sections.
In vitro differentiation
One NT-ESC line (D211) was examined further in terms of its ability to form embryoid bodies in suspension culture and its ability to undergo directed differentiation in vitro to cells representative of all three germ layers. For embryoid bodies formation, ESC colonies were cut into pieces and cultured in suspension in DMEM/Ham F-12 or RPMI media (both from Invitrogen) supplemented with 2% FBS (Hyclone, Logan, UT, USA) and 55 μM 2-mercaptoethanol in ultralow attachment six-well plates (Corning, Corning, NY, USA) for 21–28 days. To assess directed differentiation potential of NT-ESC they were plated onto fresh feeder layer and cultured for 5–6 days in ESC medium and then differentiated using modified protocols described previously (Vassiliev et al., 2010b) for ectoderm, mesoderm, and endoderm differentiation.
For ectoderm differentiation the ES medium was replaced with DMEM/F12 (1:1) medium containing 2% SR, 1× N2 supplements, 55 μM 2-mercaptoethanol, and 10 ng/mL recombinant human basic FGF (all from Invitrogen), and colonies were cultured for another 28–35 days. Resultant colonies where examined morphologically and then fixed with 4% paraformaldehyde and examined with immunofluorescence analysis for neuro-ectodermal differentiation by staining for glial fibrillary acidic protein (GFAP), nestin, and rhodopsin. For GFAP colonies were stained with primary mouse anti-GFAP monoclonal antibodies (1:100; Millipore) and rabbit antimouse IgG-Alexa 488 secondary antibodies (1:200; Invitrogen). For nestin colonies were stained with primary goat antinestin polyclonal antibodies (1:100; Santa Cruz) and donkey antigoat IgG-Alexa 647 (1:200; Invitrogen) secondary antibodies. For rhodopsin colonies were stained with primary mouse antirhodopsin monoclonal antibodies (1:100; Millipore) and rabbit antimouse IgG-Alexa 488 secondary antibodies (1:200; Invitrogen).
To induce endoderm and mesoderm differentiation NT-ESC colonies were initially cultured for 5 days in porcine ES cell medium in the presence of 100 ng/mL human recombinant Activin A (R&D Systems). For mesoderm differentiation the porcine ES cell medium was replaced with Iscove medium containing 2% FBS (Hyclone), 1 μM dexamethasone, 50 μg/mL ascorbic acid, 450 μM thioglycerol (all from Sigma) 1× ITS and 1× of penicillin/streptomycin solutions (both from Invitrogen), and colonies were cultured for 28–35 days. At the end of culture period, colonies were fixed with 4% PFA and stained with mouse anti-titin monoclonal antibodies (1:100; Sigma) and rabbit antimouse Alexa-488 antibodies (1:500; Invitrogen).
For endoderm differentiation ES culture medium was replaced with RPMI medium supplemented with 2% SR, 10 μM Nicotine amide, 10 nM exendin 4 (both from Sigma) and 50 ng/mL insulin growth factor II (IGF-II; Invitrogen), and colonies cultured for another 35–45 days. After culture, colonies were fixed and expression of pancreatic and duodenal homeobox 1 protein (PDX-1) examined with rabbit anti-PDX1 polyclonal antibodies (1:500) and donkey anti-goat IgG-Alexa 488 secondary antibodies (both from Millipore).
Imaging analysis was done using a confocal laser microscope SP5 (Leica) using Z-stack sectioning with 0.4 μm sections.
Production of porcine chimaeric blastocysts
Colonies from the D211 NT-ESC line were treated for 2–3 min with TrypLE Express to produce single cells or small clumps of cells (four to sixcells), which were washed twice in HEPES-buffered NCSU23 containing 10% FBS and then resuspended in 100 μL drops of the same medium. Twenty microliter drops from these suspensions were then placed under mineral oil on 35-mm Petri dish lids. NT-ESCs were stained with 10 nM SYTO 64 red fluorescent nucleic acid stain (Invitrogen) and injected into the blastocoele of day 5 host blastocysts. On the following day incorporation of injected ES cells into ICM of host blastocysts was examined using fluorescence TS 100 microscope (Nikon, Japan).
Statistics
The effect of TSA treatment on day 6 NT blastocyst development and primary outgrowth formation was analyzed using a one-way chi-square test for small samples using GraphPad Prizm 5 software (GraphPad Software, LaJolla, CA, USA). A value of p < 0.05 was considered to be statistically significant. Experiments were replicated at least four times and the data pooled.
Results
Experiment 1. Effect of TSA treatment on NT blastocyst development and embryonal outgrowth formation
The effect of TSA treatment on the in vitro development of porcine NT embryos and the establishment of primary outgrowths is shown in Tables 1 and 2. Treatment of NT embryos for 24 h with TSA increased the percentage of embryos that developed to the blastocyst stage (Table 1) compared with controls (15 vs. 30%; p < .001). However, there was no difference in the efficiency with which embryonal outgrowths could be established from TSA treated blastocysts (Table 2) compared with controls (18 vs. 19%, respectively; p > .05). Following plating the trophectoderm cells began to detach after 1 day and the rest of cells started to form primary outgrowths within 4–6 days after plating. These outgrowths formed colonies that were morphologically homogenous and grew as a monolayer. Outgrowths consisted of cells that displayed a morphology the same as that observed previously for ESCs isolated from IVP embryos (Vassiliev et al., 2010a) namely a polygonal shape, 12–15 μm in diameter with large nuclei to cytoplasm ratio with many visible lipid inclusions. After 8–13 days colonies had grown to 1.5–5 mm in diameter (Fig. 1A). In both groups embryonal outgrowths expressed Oct-4 and Nanog (Fig. 2A, C, and E), but did not express Gata-6 and Cdx-2.

Morphology of primary embryonal outgrowths and putative porcine ESC lines developed from cloned porcine embryos. Figure shows representative morphology of primary embryonal outgrowths developed from TSA-treated NT embryos (
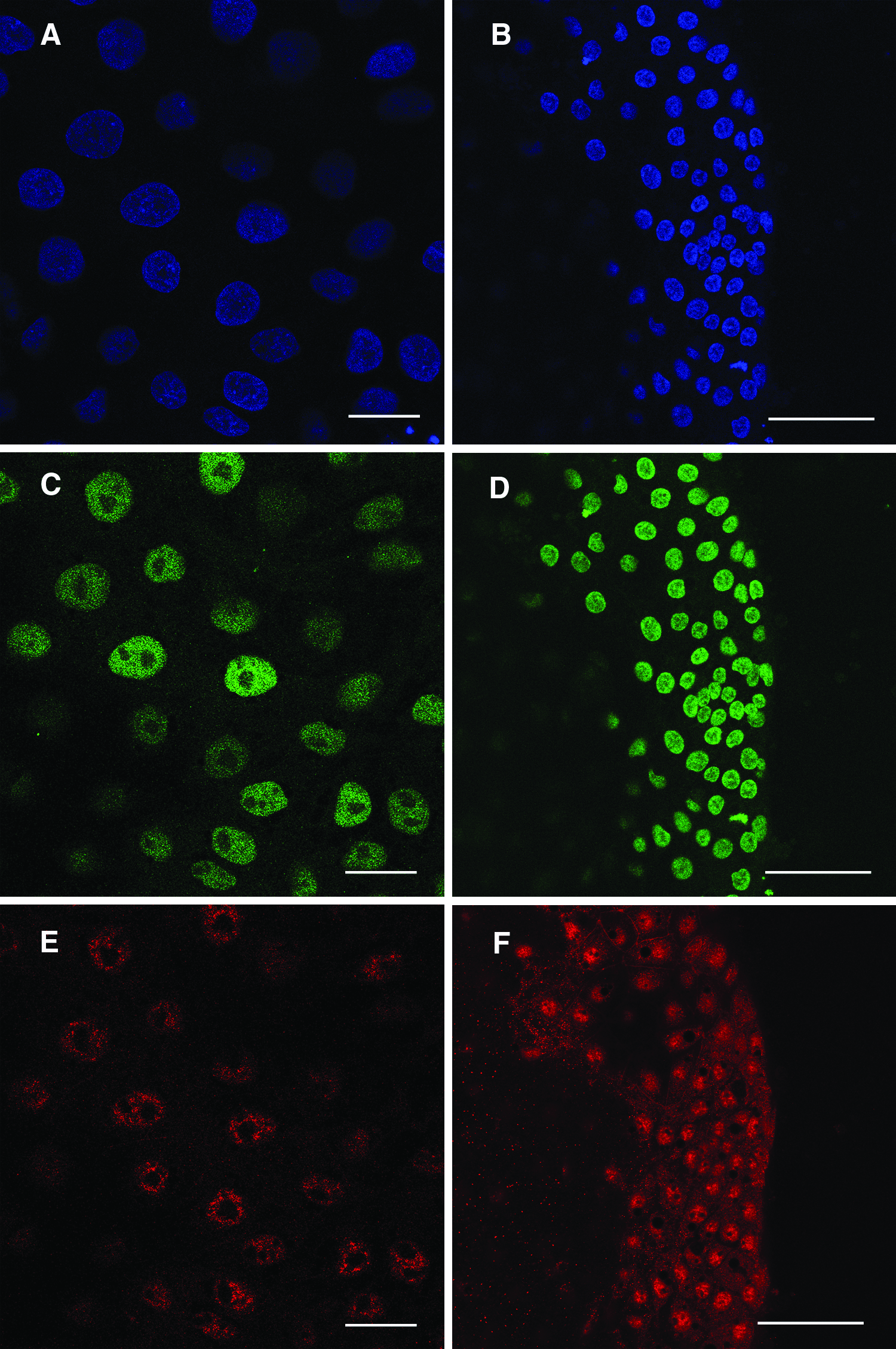
Expression of markers of pluripotency in primary embryonal outgrowths developed from TSA-treated and TSA nontreated embryos and a putative porcine ESC colony developed from from TSA-treated SCNT embryos. Both embryonal primary outgrowths (
Values are significantly different (p < 0.001).
Five replicates.
Six replicates.
There was no significant difference between treatments. Data is from four replicates. (p > 0.05).
Experiment 2. Establishment and characterisation of porcine putative NT-ESC lines from TSA-treated NT embryos
The effect of TSA treatment on the isolation of putative NT-ES cell lines is shown in Table 3. Eight primary embryonal outgrowths were obtained after plating 45 NT control embryos (17.7%). Of these none survived after two passages. As in Experiment 1, TSA-treated NT embryonal outgrowths started to develop 4–6 days after plating onto MEF layers. Over the next 8–10 days these grew in size to 2–5 mm in diameter without changes in their morphology. At this stage outgrowths were passaged onto fresh feeder layers to establish putative NT-ESCs lines or were examined for pluripotent marker expression. Of the eight primary outgrowths obtained from 46 TSA-treated NT embryos five (10.9% of plated embryos) survived repeated passaging and vitrification and continued to grow without any changes in their morphology, karyotype, and marker expression. One of these lines (D211) was then characterized further. This line continued to grow and maintain its morphology to passage 15, during which it was vitrified eight times (Fig. 1B) and exhibited a stable karyotype (Fig. 1C). This endpoint was arbitrarily chosen prior to the commencement of the experiment and we are yet to determine how long these cells can be cultured. Immunofluorescence analysis at passage 15 demonstrated that these colonies consisted entirely of cells which expressed Oct4 and Nanog (Fig. 2B, D, and F). These cells were also negative for Gata-6 and Cdx-2.
Data is from four replicates.
NT-ESCs formed embryonal bodies in suspension culture and could be directed to differentiate to cell types representative of all three germ layers. Long-term culture for 28–35 days in N2 supplement containing the neural differentiation agent putrescine, produced neural progenitors that expressed nestin (early marker of neural differentiation) and/or GFAP (marker of glial cells). Coexpression of GFAP and nestin in some of these cells indicated that these may have differentiated further to glial lineages (Fig. 3A and B). These cells also expressed β-tubulin III and rhodopsin (a pigment of retina responsible for formation photoreceptor cells in eye development) (Osakada et al., 2009). Following long-term culture for 35–45 days in mesoderm differentiation medium, titin-positive cells were found (Fig. 3C and D). This has been used as an early marker of mesoderm as it is an important component of developing striated muscles such as skeletal or cardiac muscle (van der Loop et al., 1996).
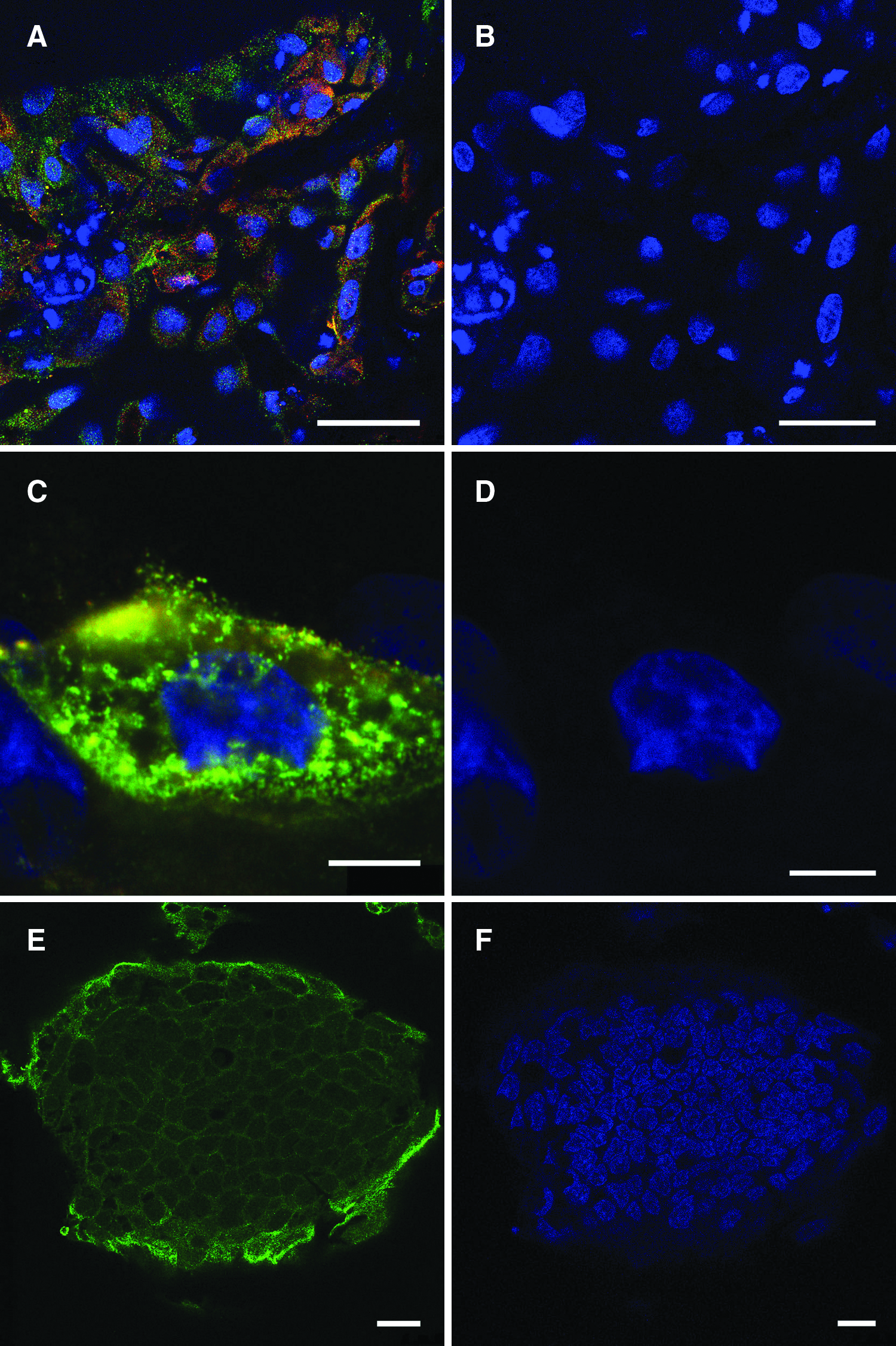
In vitro differentiation of putative porcine ESCs isolated from cloned embryos. Directed differentiation of putative porcine NT-ESCs to ectoderm resulted in the formation of various neuroectodermal cell types. After long-term culture in neuronal differentiation medium ESC colonies were stained (
These cells could also be differentiated to pancreatic islet progenitor cells. ESCs changed their morphology to endoderm-like cells during the initial endoderm differentiation step when cultured in presence of Activin A. During the next 21 days these cells grew as a monolayer and started to form dome-like clusters resembling islets. Both endoderm-like cells, which continued to grow as a monolayer, and these cell clusters expressed the pancreatic and duodenal homeobox 1 protein PDX-1 (Kahan et al., 2003) (Fig. 3E and F). Staining was localized in cytoplasm. This may have been a consequence of the relatively low concentration of glucose in RPMI medium used for endoderm differentiation as PDX-1 is normally localized in the nucleus, however, when cells are cultured in media containing low amounts of glucose PDX-1 localizes in the cytoplasm (Macfarlane et al., 1999). Injection of D211 cells labeled with DNA tracer SYTO 64 at passage 10 into the blastocyst cavities of day 5 host blastocysts resulted in the majority of these cells becoming localized in the inner cell masses of host blastocysts (Fig. 4A and B).
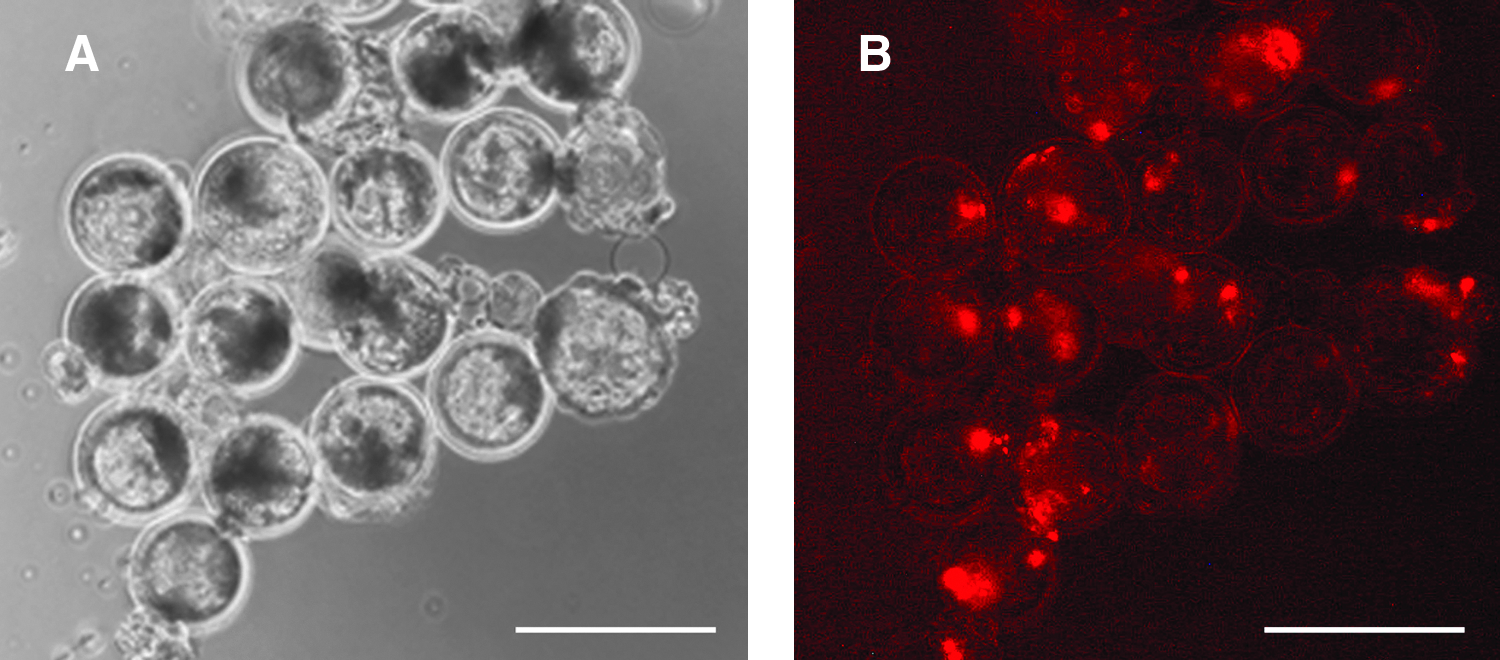
In vivo developmental potential of porcine NT-ESCs from line D211. After dissociation cells were labeled with Cyto 64 red fluorescent DNA tracer and injected into day 5 in vitro produced blastocysts. Examination next day using fluorescence revealed that the injected cells preferentially localized in the inner cell mass.
Discussion
The aim of the present study was to determine whether ESCs can be isolated from cloned embryos in the pig. To date, there have only been a few reports regarding the isolation of ESC from cloned embryos notably in mice, cattle, and primates (Byrne et al., 2007; Kawase et al., 2000; Munsie et al., 2000; Wakayama et al., 2005; Wang et al., 2005). However, at the time we undertook this study there had been no reports for the pig. We have recently reported a new approach for the isolation of porcine ESC from IVP and in vivo embryos including the production of chimaeric pigs (Vassiliev et al., 2010a, 2010b). Here we report the use of this method for isolation of putative porcine ESCs from porcine NT embryos treated with TSA. Ttichostatin A was used in the present study because we have previously shown that this can increase the number of cloned embryos that develop to the blastocyst stage as well as their total cell number (Beebe et al., 2009b). Moreover previous work in mice had shown that TSA can increase the efficiency with which ES cells can be derived from cloned embryos (Kishigami et al., 2006). In the present study more blastocysts were obtained in the TSA treatment group compared with the control group. Furthermore, putative NT-ESC lines were isolated only from this group. The reasons for this difference are unclear. At the same time the present study was undertaken Kim et al. (2010) reported the isolation of two porcine NT-ESC lines from two fetal fibroblast primary cultures at efficiencies of 2.2 and 7.1%. This is lower than the that obtained in the present study, suggesting that TSA can increase the efficiency with which NT-ESC lines can be isolated in the pig. However, further work is required to determine this. Furthermore, these workers did not freeze these lines, which is considered an essential step in the establishment of a cell line and which makes any meaningful comparisons difficult.
As described previously by us for ESCs derived from IVP and in vivo-derived embryos embryonal outgrowths from TSA-treated NT embryos were homogenous with all cells expressing Oct-4 and Nanog (Fig. 2A, C, and E). Furthermore, none of these expressed Gata-6 and Cdx-2, confirming that these outgrowths did not contain cells of extraembryonic endoderm or trophoblast origin. Of the 46 TSA-treated cloned embryos plated five cultures of primary embryonal outgrowths (10.9%) survived repeated passaging and vitrification and continued to grow without any changes in their morphology, karyotype, and marker expression. This efficiency was similar to that observed previously by us for ESC lines derived from in vivo embryos (Vassiliev et al., 2010a). NT-ESCs had a similar morphology to that described previously by us for ESCs derived from IVP and in vivo porcine embryos (Vassiliev et al., 2010b), namely, polygonal in shape, a relatively small (10–15 μm) cell diameter and a small cytoplasmic/nuclear ratio with nuclei having multiple nucleoli and multiple lipid inclusions in the cytoplasm. These cells continued to express Oct4 and Nanog, did not express Gata-6 and Cdx-2 and maintained their karyotype up to 15 passages. This was an arbitrary endpoint chosen prior to the commencement of the experiments, and we are yet to determine how long our ESCs can be passaged for. However, this was two to three times longer than fibroblasts can be cultured which, for example, may allow multiple rounds of gene targeting to be performed. In highlighting this it should also be mentioned that no ESC lines are truly immortal and loose their ability to produce chimaeras over time (Federov et al., 1997; Li et al., 2007) as well as accumulate chromosomal errors (Hughes et al., 2007). Hence, the ability to regenerate these line by isolating ES cells from cloned embryos may provide a way of overcoming this. NT-ESCs formed embryonal bodies and could be directed to differentiate to derivatives of all three germ layers. As well, when injected into blastocysts these cells preferentially localized in the ICM of host blastocysts. Further work is required to confirm the pluripotency of these cells. In particular, we need to produce chimaeric pigs to determine their ability to contribute to all the cell types in the body including the germ cells as per the original definition developed in mice. Once we have confirmed their pluripotency these cells could be used for a variety of applications. For instance, this approach will allow ESCs to be derived from animals that have a valuable genotype including males. This method may also allow the pig to be used as a large animal model for human therapeutic cloning. In particular, the ability to generate large numbers of cloned embryos relatively inexpensively will allow a range of studies to be undertaken, which so far have proved prohibitive due to the relatively low numbers of human oocytes available and the ethical considerations that surround their use.
In conclusion, we have isolated and characterized in vitro putative porcine ESCs from cloned porcine embryos treated with 50 nM TSA. These cells are similar to that described previously by us previously for IVP and in vivo-derived porcine embryos. These cells have the potential to be used for a variety of applications including as a large animal model for therapeutic cloning. However, further work is needed to confirm the pluripotency of these cells in vivo including the production of chimaeric pigs and the demonstration of germ line transmission
Footnotes
Acknowledgments
We thank Rhonda Hutchinson and Sharon Bain from Cytogenetics, Genetics and Molecular Pathology (SA Pathology at Women's and Children's Hospital, Adelaide, South Australia) for help with the karyotype analysis. This work was funded by an JDRF program project grant (4-2006-1028) to M.B.N.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.