
Abstract
Abstract
Human embryonic stem (ES) cells are usually maintained in the undifferentiated state by culturing on feeder cells layers of mouse embryonic fibroblasts (MEFs). However, MEFs are not suitable to support human ES cells used for clinical purpose because of risk of zoonosis from animal cells. Therefore, human tissue-based feeder layers need to be developed for human ES cells for clinical purpose. Hereof we report that human amniotic mesenchymal cells (hAMCs) could act as feeder cells for human ES cells, because they are easily obtained and relatively exempt from ethical problem. Like MEFs, hAMCs could act as feeder cells for human ES cells to grow well on. The self-renewal rate of human ES cells cultured on hAMCs feeders was higher than that on MEFs and human amniotic epithelial cells determined by measurement of colonial diameters and growth curve as well as cell cycle analysis. Both immunofluorescence staining and immunoblotting showed that human ES cells cultured on hAMCs expressed stem cell markers such as Oct-3/4, Sox2, and NANOG. Verified by embryoid body formation in vitro and teratoma formation in vivo, we found out that after 20 passages of culture, human ES cells grown on hAMCs feeders could still retain the potency of differentiating into three germ layers. Taken together, our data suggested hAMCs may be safe feeder cells to sustain the propagation of human ES cells in undifferentiated state for future therapeutic use.
Introduction
Human amniotic membrane is usually regarded as postlabor medical waste. This property can avoid quite some ethical problems against clinical application of amniotic membrane and its derivatives. Human amniotic membrane includes a large amount of human amniotic mesenchymal cells (hAMCs) and human amniotic epithelial cells (hAECs). HAMCs are derived from embryonic mesoderm, while hAECs are derived from embryonic ectoderm. More than 107 hAMCs could be isolated from one single amnion by simple enzyme digestion procedures. HAMCs were easily expanded in vitro for at least 15 passages without any visible morphological alterations (Alviano et al., 2007). HAMCs have great advantage for clinical usage on account of their safety property. They do not express telomerase. This can exclude risk of tumor formation after cell transplantation (Bilic et al., 2008). Those overall properties make hAMCs ideal cell resource of feeders for stem cell culture. In this article, we reported a protocol of using hAMCs isolated from full-term amniotic membrane to sustain propagation of human ES cells in undifferentiated state comparing with that on MEFs. Moreover, hAMCs may make a break through in the limitation of clinical application of human ES cells. Overall, this study provides a human-source feeder cell to support human ES cells in vitro culture for further clinical applications.
Materials and Methods
Isolation of hAECs and hAMCs
Placentas were obtained at elective cesarean section with informed consent. In addition, all the correlated ethic issues concerning this study were approved by corresponding institutional review board (IRB number: CJFH001035). Amnion cells were prepared according to protocols previously described by Akle et al. (1981) with certain modifications. Amnion layer was mechanically peeled off from chorion layer and washed several times with Hanks' balanced salt solution (HBSS) without calcium and magnesium to remove blood as completely as possible. Then, the amnion was cut into pieces of 3 cm2 and digested with 0.25% trypsin (Gibco BRL, Gaithersburg, MD) at 37°C for 15 min. The first digestion supernatant was discarded. The remaining amnion was digested with 0.25% trypsin at 37°C for 30 min consecutively twice. Cells obtained from these subsequent twice digestions, mostly comprising of hAECs, were pooled and washed three times with HBSS. Viability of hAECs was determined by exclusion of trypan blue dyeing and counted with a hemocytometer. The hAECs were collected by centrifugation, washed thoroughly with phosphate-buffered saline (PBS) and cultured in DMEM (Hyclone, UT) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA).
HAMCs were further isolated from the remainder of the third digestion by incubation with 0.1% collagenase V (Sigma-Aldrich, St. Louis, MO; dissolved in DMEM/F12) at 37°C for additional 30 min followed with centrifugation (1500 rpm, 10 min) and thorough washing with PBS. Then, hAMCs were cultured in DMEM/F12 supplemented with 10% FBS. HAECs and hAMCs of no more than 3 passages were used for experiments.
H1 ES cells growth on feeder layers
106 MEF, hAMCs, or hAECs were respectively seeded onto 60-mm cell culture dishes (BD Falcon, 353002). Then they were cultured for about 2 days until they were confluent. Before being used, they were treated with 10 μg/mL mitomycin C (Sigma-Aldrich) for 1 h. H1 ES cells were dissociated by treating with DMEM/F12 containing 1 mg/mL collagenase IV (Sigma-Aldrich). The dissociated ES cells were respectively plated onto the confluent MEF, hAMCs, and hAECs feeder cells. Then the ES cells were cultured in medium consisting of DMEM/F12 with 20% knockout serum replacement (KSR, Invitrogen), 2 mM L-glutamine, 1% nonessential amino acids, 0.1 mM β-mercaptoethanol (Invitrogen), 4 ng/mL recombinant human basic fibroblast growth factor (bFGF, PeproTech, Rockhill, NJ) and 0.5% penicillin and streptomycin (Invitrogen).
Colonial diameters and growth curve examination of ES cells
ES cells (1 × 104) were respectively seeded onto hAMCs, hAECs, and MEFs feeders. Each of them was cultured with three repetitive dishes. On day 2 and day 5, every ES colonies grown up were taken photos. These are totally 15, 17, and 14 colonies grown up on MEFs, hAMCs, and hAECs dishes, respectively. Colonial diameters were measured and statistically analyzed. To determining growth curve, 1 × 106 ES cells were respectively plated onto MEFs, hAMCs, and hAECs dishes, and cell numbers were counted on days 1, 2, 3, and 4. The experiment was performed in triplicate.
Cell cycle analysis
ES cells grown on MEFs, hAMCs, or hAECs were respectively trypsinized and fixed with 70% ethanol for over 4 h. The cells were then pelleted and washed with PBS plus 20 mM EDTA. RNA was removed by incubating samples with RNase (1 mg/mL) at 37°C for at least 1 h. Cells were then stained with 30 μg/mL propidium iodine. DNA content of at least 10,000 cells per sample was analyzed by fluorescence-activated cell sorter (FACS, Becton Dickinson FACS Calibur, excitation wavelength: 488 nm). Cell cycle was analyzed using FlowJo software (ver 5.7.2).
Immunofluorescence cytochemistry
ES cells or hAMCs were washed with PBS and fixed with 4% paraformaldehyde for 20 min at room temperature (RT). The fixative solution was removed and the cells were rinsed three times with PBS. The cells were processed with respective primary antibodies at 4°C overnight, anti-Oct3/4 (mouse monoclonal, Abcam), anti-SOX2 (rabbit polyclonal, Santa Cruz, Santa Cruz, CA), anti-NANOG (mouse monoclonal, R&D, Indianapolis, IN), anti-SSEA4 (mouse monoclonal, Abcam, Cambridge, MA), anti-TRA-1-81 (mouse monoclonal, Abcam), anti-TRA-1-60 (mouse monoclonal, Abcam), anti-CK19 (mouse monoclonal, Abcam), antivimentin (goat polyclonal, Santa Cruz). Then cells were incubated with corresponsive secondary antibodies conjugated to Fluorescein (FITC, Santa Cruz) or rodamine (Santa Cruz). After washing, coverslips were mounted and digital images were acquired with microscope.
Fluorescence-activated cell sorter analysis
HAECs were collected by trypsin digestion and centrifugation. Then hAECs were washed three times with PBS. Specific primary monoclonal antibody for epithelial marker cytokeratin 19 (CK19; Santa Cruz, sc-6278, 2 μg/mL) was used. Isotype immunoglobulin was used as negative control. (Santa Cruz, sc-3878). Fluorescein isothiocyanate (FITC)-labeled goat antimouse IgG (Santa Cruz, sc-2010) was used as secondary antibody. Cells were prepared at 1 × 106 cells/mL in PBS and were analyzed on flow cytometer (Becton Dickinson FACS Calibur, Franklin Lakes, NJ). A minimum of 10,000 events was acquired for each sample.
Preparation of cell lysates and immunoblotting
Cells were lysed in 10 mM Tris (pH 7.4), 1 mM EDTA, 0.5 mM EGTA, 150 mM NaCl, 1% Triton X-100, 50 mM NaF, 10 mM Na4P2O7 · 10 H2O, 5 μg/ml aprotinin, 5 μg/mL leupeptin, and 1 mM PMSF. Aliquots (50 μg) of protein were electrophoresed on SDS-polyacrylamide gels and transferred onto nitrocellulose membranes (Hybond™ ECL). The membranes were blocked with 5% skim milk in Tris-buffered saline (TBS) containing 0.1% Tween-20 (TBS-T) and subsequently incubated overnight at 4°C with following primary antibodies (1:3000 dilution): anti-CK19 (mouse monoclonal, Abcam), anti-Ecad (mose monoclonal, Sigma), anti-β-actin (mouse monoclonal, Sigma), anti-α-SMA (mouse monoclonal, Sigma); anti-Oct3/4 (mouse monoclonal, Abcam), anti-SOX2 (rabbit polyclonal, Santa Cruz), anti-NANOG (mouse monoclonal, R&D). After washing with TBS-T for 1 h at room temperature, the membranes were further incubated with corresponding horseradish peroxidase-conjugated secondary antibodies (Santa Cruz) for 2 h, followed by 45 min of washing (with three to five changes of the washing buffer). Protein bands were visualized with Super Signal Reagents (Pierce, Thermo Fisher Scientific, Rockford, IL).
Embryoid body-mediated in vitro differentiation
For EB formation, human ES cells were harvested by digestion with collagenase IV. The clumps of the ES cells were transferred to poly (2-hydroxyrthyl methacrylate)-coated Petri dish (BD Falcon, Bedford, MA) in DMEM/F12 containing 20% knockout serum replacement (KSR, Invitrogen), 2 mM L-glutamine, 1% nonessential amino acids, 0.1 mM β-mercaptoethanol (Invitrogen), and 0.5% penicillin and streptomycin. The medium was changed every other days. After 8 days of floating culture, EBs were collected by centrifugation.
mRNA extraction and RT-PCR
Total RNA was extracted from human ES cells and EB using Trizol Reagent. Then mRNA was reversely transcribed at 42°C for 30 min using ReverTra Ace-α- (Toyobo). The primers for PCR amplification of markers of three germ layers are listed in Table 1.
Teratoma formation
Human ES cells were harvested with collagenase IV treatment. They were collected into tubes for centrifugation. The pellets were suspended in DMEM/F12. Human ES cells (1 × 106) were injected into hind leg of severe combined-immunodeficient (SCID) mice. Eight weeks after injection, tumors were dissected and fixed with PBS containing 4% paraformaldehyde. Paraffin-embedded tissues were sliced and stained with hematoxylin and eosin.
Statistical Analysis
Quantitative data are expressed as mean ± SD. Statistical significance was determined by two-tailed Student's t-test or one-way ANOVA followed by the LSD-t test for multiple comparisons. A p-value of less than 0.05 was considered statistically significant.
Result and Discussion
Isolation of hAMCs and hAECs from human amniotic membrane
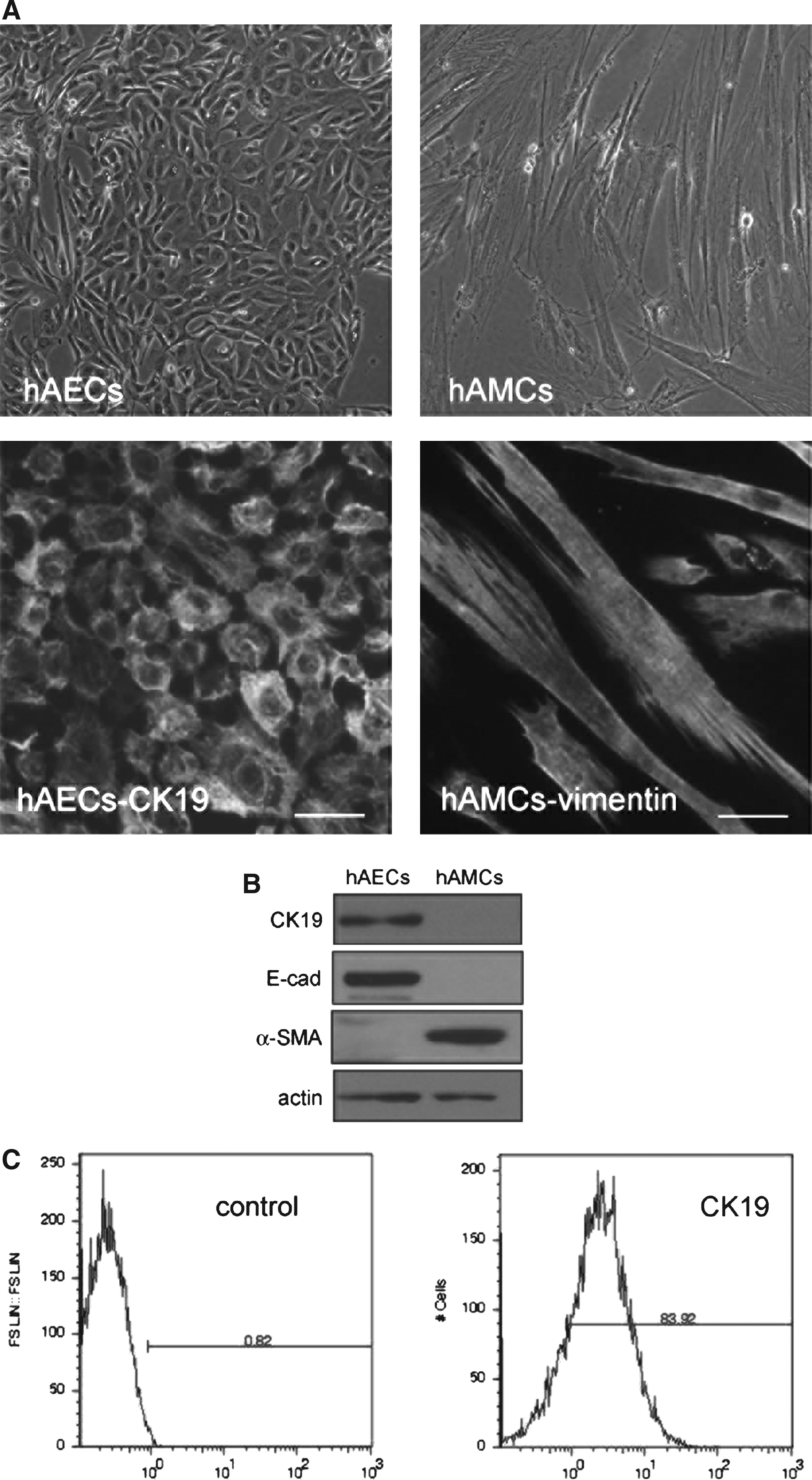
HAECs and hAMCs could be isolated by digestion with trypsin and collagenase V, respectively, through accurate control of digestion time and enzyme concentration. Primary hAECs exhibited typical epithelial morphology with tight cell–cell contact and expressed epithelial maker cytokeratin 19 (CK19), whereas hAMCs exhibited typical mesenchymal morphology with bulking cell body and spindle-like cell shape and mesenchymal marker vimentin (Fig. 1A). Epithelial markers such as CK19 and E-cadherin could be detected in hAECs but not in hAMCs. On the contrary, mesenchymal marker α-smooth muscle actin could only be detected in hAMCs (Fig. 1B). Flow cytometry analysis showed that more than 80% cells are Cytokeratin 19 positive in hAECs (Fig. 1C).
(
Morphology and growth of human ES Cells on hAMCs and hAECs
Amnion, part of placenta, is derived from epiblast during early embryonic development. It supplies physiological environment for embryonic development and fetal growth. For this reason, the amnion-derived cells could be favorable candidate for supporting ES cells' culture. In order to determine which one of hAMCs or hAECs could act as better feeder cells to support human ES cells' culture, we respectively seeded human H1 ES cell clones (Levenstein et al., 2006) on freshly isolated hAMCs or hAECs, which were pretreated with mitomycin C to inhibit their proliferation. We found that H1 ES cells formed tightly packed flat colonies with clear boundaries on both hAMCs and hAECs. The morphology of ES cells grown on either hAMCs or hAECs was similar to that grown on MEFs, characterized by large nuclei and scant cytoplasms (Fig. 2A). However, we observed that ES colonies on hAECs are obviously smaller than those on hAMCs or MEFs. We further measured the colonies' diameters after respectively seeding the same number of human ES cells onto MEFs, hAMCs, or hAECs as feeder cells. The results showed that diameters of human ES colonies on hAMCs were slightly bigger than that on MEFs, whereas the colonies diameters of human ES on hAECs were significantly the smallest (Fig. 2B).

Morphology and colonial diameters of human ES Cells grown on hAMCs, hAECs, and MEFs. (
In order to determine the proliferating capacity of ES cells respectively cultured on hAMCs, hAECs, or MEFs, 1 × 106 ES cells were plated in triplicate culture dishes, and cell numbers were counted on days 1, 2, 3, and 4. The ES cells' proliferation rates were almost equal in cultures on hAMCs and MEFs, and were higher compared with that on hAECs (Fig. 2C). Flow cytometry was performed to analyze DNA content in ES cells respectively cultured on MEFs, hAMCs, or hAECs. Results showed that cell cycle distributions in ES cells grown on hAMCs were the same as those of ES cells grown on MEFs. In contrast, the ES cells in S phase grown on hAECs were fewer than those grown on hAMCs or MEFs (Fig. 2D).
hAMCs maintain pluripotency of human ES cells
HAECs isolated from human full-term placenta comprised a certain subpopulation that expressed pluripotent markers such as Oct-3/4, Sox2, and SSEA-4 (Miki et al., 2005), indicating that hAECs can provide appropriate environment for maintaining undifferentiated state of stem cells. It has been reported that hAECs could maintain the pluripotency of human ES cells (Lai et al., 2010; Miyamoto et al., 2004) as well as mouse ES cells (Lai et al., 2009). Like hAECs, hAMCs also comprised stem cell-like cells and various progenitor cells, which could differentiate into many types of mature cells such as osteoblast, cartilage cell, adipocyte, cardiomyocytes, vascular endothelial cells, and neural cells (Diaz-Prado et al. 2010, 2011; Kim et al., 2007; Moon et al., 2008). HAMCs also expressed well-defined human mesenchymal stem cell markers such as CD90, CD73, CD166, CD105, CD44, and CD29, as well as embryonic stem-cell markers like Oct-3/4, SSEA-3, -4, and STRO-1 (Bilic et al. 2008; Diaz-Prado et al. 2010). In order to assess whether hAMCs could support pluripotency of human ES cells as well as other feeder cells, we investigated the expression of several stem cell markers on human ES cells. Immunofluorescence staining showed expression of Oct-3/4, Sox2, Nanog, SSEA-4, TRA-1-81, and TRA-1-60 on human ES cells grown on hAMCs for 10 passage (Fig. 3A). Immunoblotting showed that Oct-3/4, Sox2 and Nanog were highly expressed in human ES cells grown on hAMCs or hAECs, as well as on MEFs (Fig. 3B).

Pluripotency of human ES cells grown on hAMCs. (
Developmental potential of human ES cells grown on hAMCs
In order to evaluate the in vitro differentiation ability of human ES cells grown on hAMCs, we checked embryoid bodies' (EBs) formation in floating culture of human ES cells after being passaged for 20 times on hAMCs. After 8 days in suspension culture, human ES cells formed ball-shaped structures (Fig. 4A). RT-PCR detected that these EBs expressed markers of endoderm such as forkhead box A2 (FOXA2), AFP, cytokeratin 8 and 18, SRY-box containing gene 17 (SOX17), markers of mesoderm such as α-smooth muscle actin (α-SMA), Msh homeobox1 (MSX1), markers of ectoderm such as microtubule-associated protein 2 (MAP2), and paired box 6 (PAX6) (Fig. 4B). In contrast, expression of Oct-3/4 was markedly decreased. These data demonstrated that iPS cells could differentiate into three germ layers in vitro.
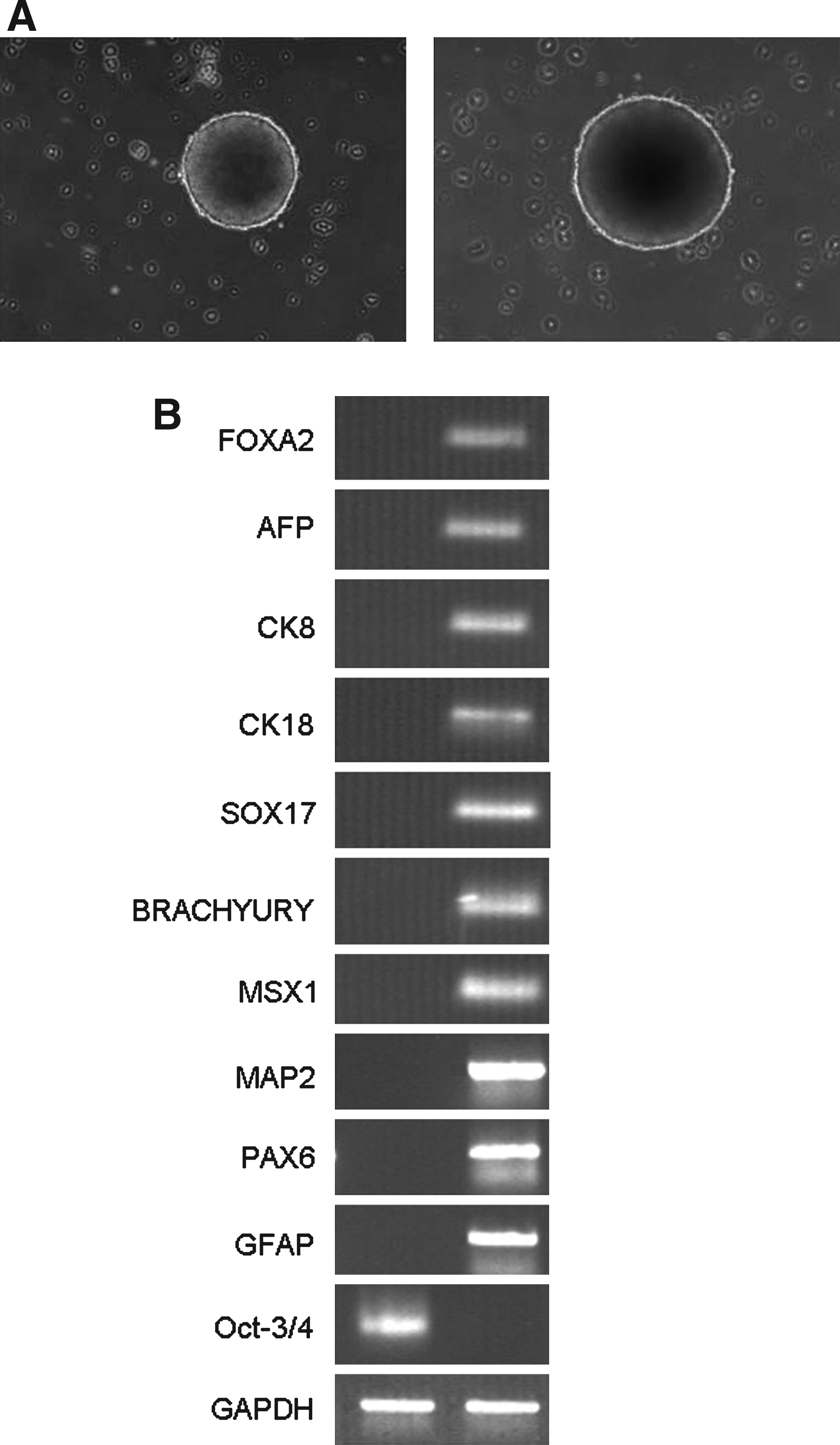
Embryoid body-mediated differentiation of human ES cells grown on hAMCs. (
In order to test developmental pluripotency in vivo, we transplanted 1 × 106 human ES cells cultured on hAMCs into hind leg of SCID mice. Eight weeks after injection, we observed teratomas' formation. Histological examination showed that the tumors contained various tissues (Fig. 5), including muscle (mesoderm), adipose tissue (mesoderm), bone (mesoderm), cartilage (mesoderm), neural tissue (ectoderm), keratin-containing epidermal tissues (ectoderm), pigmented epithelium (ectoderm), sebaceous glands (ectoderm), and intestinal epithelium (endoderm). Various tissues of three germ layers were found in teratomas, containing integrated skin and its accessories (cornified epidermis, hair follicles, sebaceous glands), immature uterine endometrium-like glands, alveolus-shaped epidermis, indicating good developmental potential of the human ES cells cultured on hAMCs. These overall indicated that hAMCs could maintain human ES cells in undifferentiated state with good developmental potentials, and suggested hAMCs was ideal feeder cells for ES cells' culture.

Teratoma-derived from human ES cells cultured on hAMCs. Human ES cells were cultured for 20 passages on hAMCs. Hematoxylin and eosin-stained sections of teratomas derived from SCID mouse after 8 weeks of transplantation were shown. The teratomas contained all three embryonic germ layers. Scale bars: 50 μm.
Taken together, this study appraised properties of certain novel feeder cells originated from routinely discarded human amnion tissues to sustain in vitro growth and maintain undifferentiated state of human ES cells. This protocol might minimize ethic problem of human ES cells for future therapeutic usage.
Footnotes
Acknowledgments
We thank Dr. Qingsheng Yu and Dr. Xiaoqing Qiu (West China Hospital, Sichuan University, Chengdu, China) for critically reading of this manuscript. This work was supported by grants from the Chinese National 863 Project (2006AA02A114), Beijing Ministry of Science and Technology (D07050701350704).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
Supplementary Data
See online Supplementary data at ![]() .
.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.