
Abstract
Abstract
Human-induced pluripotent stem cells (hiPSCs) are expected to become a powerful tool for regenerative medicine. Their efficacy in the use of clinical purposes is currently under intensive verification. It was reported that hiPSC-derived hemangioblasts had severely limited expansion capability due to an induction of early senescence: hiPSC-derived vascular endothelial cells (VECs) senesced after one passage and hiPSC-derived hematopoietic progenitor cells (HPCs) showed substantially decreased colony-forming activities. Here we show that early senescence is not an inevitable fate of hiPSC-derived cells. Applying our unique feeder-free culture methods for the differentiations of human embryonic stem cells (hESCs), we successfully generated VECs and HPCs from three lines of hiPSCs that were established by using a retrovirus vector system. All hiPS-derived VECs could be subcultured by 2:1∼3:1 dilutions up to 10∼20 passages, after which the cells underwent senescence. Among the three lines of hiPSCs, two lines generated HPCs that bore comparable granulocyte colony-forming units to those of hESCs. Moreover, one line effectively reproduced HPCs within the sac-like structures, the fields of in vitro hematopoiesis, as in the case of hESCs. Surprisingly, release of neutrophils into culture supernatant persisted even longer (∼60 days) than the case of hESCs (∼40 days). Thus, the problem of early senescence can be overcome by selecting appropriate lines of hiPSCs and applying proper differentiation methods to them.
Introduction
So far, two major issues have been raised concerning the disadvantageous outcomes of iPSCs after differentiation. One is a widely recognized matter of tumorigeneity, which was reported in the cases of miPSC chimeric mice (Okita et al., 2007) and the mice transplanted with miPSC-derived differentiated cells (Nelson et al., 2009). Already, several measures have been proposed to overcome this problem: an application of nonviral vectors (Okita et al., 2008; Yu et al., 2009), excisions of integrated transgenes (Kaji et al., 2009; Woltjen et al., 2009), a usage of L-myc instead of c-myc transgene (Nakagawa et al., 2008, 2010), and a selection of iPSC lines with least tumorigeneity after differentiation (Miura et al., 2009). Those strategies, along with the recent technological innovations to introduce the reprogramming factors via RNA-based (Fusaki et al., 2009; Seki et al., 2010; Warren et al., 2010) or protein-based systems (Kim et al., 2009; Zhou et al., 2009), are expected to make a great contribution toward the complete resolution of the problem.
The second issue is rather a newly proposed one: an induction of early senescence in hiPSC-derived differentiated cells (Feng et al., 2010). It was reported that every hiPSC established by using a retrovirus vector system suffered from expansion deficiency after differentiation into vascular endothelial cells (VECs), hematopoietic progenitor cells (HPCs), and retinal pigmented epithelium cells (Feng et al., 2010). Although the molecular mechanism of early senescence remains elusive, the effects of p53 inactivation, LIN28 activation and insertion of proviral transgenes into chromosomes were discussed (Feng et al., 2010). The finding of early senescence is worth reporting in that it has provoked a caution that we should o use during our researches on hiPSCs. Nevertheless, we thought that their conclusion must be further validated in other situations, where differentiation processes are performed by distinct methods, because early senescence is often caused by cellular stresses and the degrees of stresses substantially vary depending on culture conditions.
We previously established feeder-free methods for neutrophilic (Saeki et al., 2009) and vascular endothelial (Nakahara et al., 2009) differentiations of hESCs. By applying those methods to hiPSCs, we studied the incidence of early senescence during the differentiation of hiPSCs that were established by using a retroviral vector system. Our data indicate that early senescence is not an inevitable fate of the hiPSC-derived differentiated cells even in the presence of retroviral insertions of the reprogramming transgenes. We also show that hiPSC-derived VECs undergo senescence after 10∼20 passages, as in the cases of primary human VECs, without entering crisis. Our results clearly show that the problem of early cellular senescence can be overcome by selecting appropriate lines of hiPSCs and applying proper differentiation methods to them.
Materials and Methods
Cell culture
The use of hESCs was performed in accordance with the Guidelines for Derivation and Utilization of Human Embryonic Stem Cells of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan, after approval by the institutional review board of International Medical Center of Japan (IMCJ). The hESCs (KhES-1, KhES-3) (Suemori et al., 2006) were provided by Kyoto University (Kyoto, Japan). The hiPSC were provided by either CiRA at Kyoto University (253G1 and 253 G4 (Nakagawa et al., 2008), 201B7 and 201B2 (Takahashi et al., 2007) or by National Research Institute for Child Health and Development (#25). The cells were maintained on dishes coated with γ-irradiated murine embryonic fibroblasts (MEFs) in DMEM/F12 (Invitrogen Corp., Carlsbad, CA) supplemented with 20% Knockout™ Serum Replacement (KSR) (Invitrogen Corp.), 5 ng/mL fibroblast growth factor 2 (FGF-2; Pepro Tech Inc., Rocky Hill, NJ), 1% nonessential amino acids solution (Invitrogen Corp.), 1 mM sodium pyruvate solution (Invitrogen Corp.), 100 μM 2-mercaptethanol (Sigma Chemical Co., St. Louis, MO), 2 mM L-glutamine (Invitrogen Corp.), 20 U/mL penicillin (Invitrogen Corp.) and 20 μg/mL streptomycin (Invitrogen Corp.). The cells were passed twice a week (e.g., on Tuesday mornings and Friday evenings) by a treatment with dissociation liquid, which contains 0.25% trypsin (Invitrogen Corp.), 1 mg/mL collagenase IV (WAKO Pure Chemical Industries, Osaka, Japan), 20% KSR, 1 mM CaCl2, at 37°C for 5 to 15 min and seeded at split ratios of 1:2 to 1:4 into new MEF-coated dishes. During the course of experiments, cells showed normal karyotypes.
Vascular endothelial differentiation of hESCs/hiPSCs
Differentiation was performed as previously described (Nakahara et al., 2009) with a slight modification. The hESCs/hiPSCs were detached from culture plates by using the dissociation liquid for 15 min at 37°C. The mildly dissociated hESC/iPSC clumps were cultured in a 6-cm diameter low-attachment dish (Nalge Nunc International K.K., Tokyo, Japan) to form spheres using the differentiation medium consisting Iscove's modified Dulbecco's medium (IMDM) (Sigma Chemical Co.) supplemented with 15% heat-inactivated fetal bovine serum (FBS) (PAA Laboratories GmbH, Linz, Austria), 0.1 mM 2-mercaptoethanol, 3 mM L-glutamine, 10 U/mL penicillin, 20 ng/mL vascular endothelial growth factor (VEGFA), 20 ng/mL bone morphogenetic protein 4 (BMP4), 20 ng/mL stem cell factor (SCF), 10 ng/mL FMS-related tyrosine kinase-3 ligand (Flt3-L), 20 ng/mL Interleukin 3 (IL3), and 10 ng/mL IL6. After incubation for 3 days at 37°C under a 100% humidified condition in a 5% CO2 gas incubator, spheres were subjected to adherent culture using 100 mm × 20 mm 0.1% porcine type A gelatin (Sigma Chemical Co.)-coated dishes in the differentiation medium described above. Media were changed twice a week. For passage, cells were harvested by treatment with 0.25% trypsin and 1 mM EDTA and replated at split ratios of 1:2 on new gelatin-coated dishes.
Senescence-associated (SA)-β-galactosidase assays
The 1 × 105 VECs were cultured in 6-cm culture plates. After an incubation at 37°C in 5% CO2 incubator for 4∼5 days, cells were subjected to SA-β-galactosidase assays by using Senescence Detection Kit (BioVision Research Products Inc., Mountain View, CA) according to a manufacture's guidance.
Cord formation assays
Matrigel™ Basement Membrane Matrix, phenol-Red free (Cat 356237, BD Biosciences, San Jose, CA) was loaded into the 24 multiwell dishes (95 μL/well). After the dishes were incubated for 30 min at 37°C, 1 × 104 cells per well were seeded in differentiation medium described above. Cell morphologies were observed after overnight culture under an inverted light microscope (Olympus Optical Co. Ltd, Japan).
Uptake of acetylated low-density lipoprotein (Ac-LDL)
Cells were transferred in four-well chamber slide system (Nalge Nunc International Corp., Naperville, IL). After overnight culture, cells were washed by Hank's balanced salt solution (HBSS) twice and incubated in serum-free medium containing 10 μg/mL of low-density lipoprotein from human plasma, acetylated, DiI complex (DiI Ac-LDL) (Invitrogen Corp.) for 4 h. After washing the cells by HBSS for three times, nuclei were counterstained using 10 nM of Hoechst 33342 (Sigma Chemical Co.). After washing the cells, samples were observed under the fluorescence microscope (Olympus Optical Co. Ltd).
Flow cytometry
Cells were collected by a treatment with 0.2% EDTA or Dispase (BD Biosciences). After a wash in phosphate-buffered saline (PBS), 1 × 106 cells were reacted with first antibodies on ice for 30 min. The expression level of each protein was analyzed using a FACSCalibur™ (BD Biosciences). The antibodies used were a mouse monoclonal antihuman Tie-2- allophycocyanin (APC) antibody (R&D Systems Inc., Minneapolis, MN), antihuman VEGF receptor 1 (VEGF R1)-PE antibody (R&D Systems Inc.), mouse antihuman CD45-PE (BD Biosciences), and mouse antihuman CD11b-PE (BD Biosciences). After antibody-staining procedures, cells were stained with propidium iodide (PI) (Sigma Chemical Co.), in the case of Tie-2 staining, or TO-PRO3 fluorescent dye (Invitrogen Corp.), in the cases of VEGF1, CD45, and CD11b staining, for 10 min. During analysis, dead cells were gated out as FL-2 higher fractions, in the case of PI staining, or FL4-higher fractions in the case of TO-PRO3 staining.
Immunostaining
The cells were fixed on slide glasses by using a cytospin apparatus (Cytospin 2) along with further fixation with acetone/methanol solution (1:3). The immunostaining procedure was performed as described elsewhere (Nakahara et al., 2009) with first antibody reactions using a rabbit polyclonal antihuman p16INK4A antibody (SC-20) (Santa Cruz Biotechnology Inc., Santa Cruz, CA), a mouse monoclonal antihuman p21CIP1 antibody (Santa Cruz Biotechnology Inc.), a rabbit polyclonal antihuman endothelial nitric oxide synthase (eNOS) antibody (H-159) (Santa Cruz Biotechnology Inc.), or a rabbit polyclonal antihuman von Willebrand factor (vWF) antibody (Sigma Chemical Co.), followed by second antibody reactions using Alexa Fluor® 488 chicken antimouse IgG (H+L), Alexa Fluor® 568 goat antirabbit IgG (H+L), or Alexa Fluor® 594 chicken antigoat IgG (H+L) (Invitrogen Corp.). Nuclear counterstaining was performed using 300 nM of 4′,6-diamino-2-phenylindole (DAPI).
Hematopoietic differentiation of hESCs/hiPSCs
Differentiation was performed as previously described (Saeki et al., 2009). In brief, hESCs/hiPSCs were detached with 1 mg/mL collagenase IV (Invitrogen Corp.) and transferred to a 6 cm diameter low-attachment dish (Nalge Nunc International K.K.) coated with 2-methacryloyloxyethyl phosphorylcholine in 5 mL IMDM (Sigma Chemical Co.) supplemented with 15% FBS (PAA Laboratories GmbH), 2 mM L-glutamine, 100 μM 2-mercaptethanol, 20 U/mL penicillin, and 20 μg/mL streptomycin in the presence of 20 ng/mL insulin-like growth factor II (IGF-II; Pepro Tech Inc.), 20 ng/mL VEGFA (Pepro Tech Inc.), 100 ng/mL SCF (Pepro Tech Inc.), 100 ng/mL Flt3-L (Pepro Tech Inc.), 50 ng/mL thrombopoietin (TPO; Kirin Brewery Company, Ltd., Tokyo, Japan), and 100 ng/mL G-CSF (Kirin Brewery Company, Ltd.) (Differentiation medium) at a density of 4 × 105 cells/mL. After primary differentiation for 3 days, the spheres were transferred to 10-cm diameter dish coated with 0.1% gelatin. Spheroid cells floating in the culture supernatant were collected over time and analyzed.
Colony assays
Colony assays were performed using Methocult TM GF+H4535 (Stemcell Technologies Inc., Vancouver, Canada) in accordance with the manufacturer's recommendations. In brief, 0.3 mL of cell suspension, which contained 10 cells, was mixed in 3 mL of methylcellulose solution consisting of 1% methylcellulose, 30% FBS, 1% bovine serum albumin, 100 μM 2-mercaptoethanol, 2 mM L-glutamine, 50 ng/mL SCF (Pepro Tech Inc.), 20 ng/mL interleukin 3 (IL-3; Pepro Tech Inc.), 20 ng/mL interleukin 6 (IL-6; Pepro Tech Inc.), 20 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF; Pepro Tech Inc.), 20 ng/mL G-CSF and 3 U/mL erythropoietin (Kirin Brewery Company, Ltd.) in 3.5-cm culture dishes. After 2 weeks, the number of colonies was counted. The morphology of the colonies was observed using an inverted light microscope (Olympus Optical Co. Ltd).
Reverse transcription-polymerase chain reaction (RT-PCR)
RNA was extracted from 5 × 106 cells using an RNeasy Mini Kit (Qiagen Inc., Valencia, CA) and cDNA was synthesized using a Superscript II Kit (Invitrogen Corp.) in accordance with the manufacture's protocol. The sequence of the primers used are as follows: vascular endothelial (VE)-cadherin; a forward primer 5′-TGGGCTCAGACATCCACATA-3′ and a reverse primer 5′-TCACAGTCTCCCATTGGGAAT-3′, platelet endothelial cell adhesion molecule-1 (PECAM1); a forward primer 5′-GCAAAATGGGAAGAACCTGA-3′ and a reverse primer 5′-CACTCCTTCCACCAACACCT-3′, interferon α1 (IFNA1); a forward primer 5′-GGAGTTTGATGGCAACCAGT-3′ and a revsese primer 5′-CTCTCCTCCTGCATCACACA-3′, interferon α2 (IFNA2); a forward primer 5′-GCAAGTCAAGCTGCTCTGTG-3′ and a reverse primer 5′-GATGGTTTCAGCCTTTTGGA-3′, interferon β1(IFNB1); a forward primer 5′-ATTGCCTCAAGGACAGGATG-3′ and a reverse primer 5′-AGCCAGGAGGTTCTCAACAA-3′. As a molecular maker, DNA MW Standard Marker 100 bp DNA Ladder (Takara Shuzo Co. Ltd., Shiga, Japan) was used.
Wright-Giemsa (WG) staining and special staining procedures
Viable cells in the dishes were observed directly using an inverted phase contrast light microscope (Olympus Optical Co. Ltd., Tokyo, Japan). Alternatively, the cells were fixed on glass slides using a cytospin centrifuge (Cytospin 2, SHANDON, Pittsburgh, PA), stained with WG solution (Muto Pure Chemical Co., Tokyo, Japan), and then observed using a light microscope (Olympus Optical Co. Ltd.). Double esterase staining was performed using the staining kit (Muto Pure Chemical Co.) according to the manufacturer's protocols.
Phagocytosis
hESC-derived neutrophils attracted to the lower chamber of Chemotaxel were suspended in HBSS containing 2.5% FBS and incubated at 37°C for 1 h with 5 μL zymosan (1 mg/mL) in the presence of 100 nM fMLP. Subsequently, the cells were collected using a cytospin apparatus and stained with WG solution. Phagocytosis was determined by a microscope observation.
Nitroblue tetrazolium reduction assay for respiratory burst activity
The floating cells were collected by mild centrifugation of the culture supernatant. After washing with PBS, the cells were resuspended in 1 mL RPMI 1640 (Sigma Chemical Co.) supplemented with 10% FBS containing 1 mg/mL nitroblue tetrazolium (NBT) (Nacalai Tesque Inc., Kyoto, Japan) and 100 nM fMLP for 30 min at 37°C. After washing with PBS, the cells were resuspended in 10 μL PBS and dropped onto Matsunami Adhesive Silane (MAS)-coated glass slides (Matsunami Glass Ind., Ltd. Osaka, Japan) and the formazan blue-black deposit-containing cells were observed using a light microscope (Olympus Optical Co. Ltd.).
Results
hiPSC can generate subculturable VECs
We previously established a feeder-free method for the vascular endothelial differentiation of hESCs (Nakahara et al., 2009). The unique points of our method are (1) it is a two-tired differentiation system with a sphere-forming floating culture and a subsequent attachment culture, (2) it uses multiple hematopoietic cytokines in addition to a conventionally used cytokine of VEGF, (3) it enables high-purity production of VECs without contamination by growth-competing pericytes, (4) it enables the production of VECs that can be subcultured up to 10∼20 passages (Nakahara et al., 2009). Applying this method, we performed VEC differentiation of the following lines of hiPSCs: 253G1 and 253G4, which were established from adult human dermal fibroblasts by introducing three retroviral transgenes of oct4, sox2, and klf-4 (OSK) (Nakagawa et al., 2008), 201B7 and 201B2, which were established from adult human dermal fibroblasts by introducing four retroviral transgenes of oct4, sox2, klf-4, and c-my (OSKM) (Takahashi et al., 2007) and #25, which were established from human embryonic lung fibroblasts of MRC-5 by introducing retroviral transgenes of OSKM.
Among the five lines, 253G1, 201B7, and #25 successfully generated VECs. Although cell morphologies slightly differed from one another (Fig. 1A), we could effectively expand the VECs generated from all the three lines: the hiPSC-derived VECs were subcultured by 2:1∼3:1 dilutions up to 10 passages, in the case of 201B7, or even higher, in the cases of 253G1 and #25 (Fig. 1B). Both 253G1-derived and #25-derived VECs expanded with comparable growth rates to hESCs-derived VECs, whereas the growth speed of 201B7-derived VECs was slightly lower. After 10∼20 passages, the hiPSC-derived VECs underwent senescence as demonstrated by SA-β-galactosidase assays as in the cases of hESC-derived VECs and HUVEC (Fig. 1C). In agreement with this, the expressions of senescence-associated gene products of p16INK4A and/or p21CIP1 were induced in the senesced cells (Fig. 1D). Using the cells at exponentially growing phases, we evaluated the functions and maker expressions. All the hiPSC-derived VECs showed high Ac-LDL-uptaking capacities (Fig. 2A) and cord-forming activities (Fig. 2B). Although the expressions of VE-cadherin and PECAM1 messages were hardly detectable in #25-derived VECs (Fig. 2C), these cells showed comparable protein expressions of eNOS (Fig. 2D), Tie-2 (Fig. 2E), and VEGFR1 (Fig. 2F) to the other hiPSCs-derived VECs and HUVEC.

The VEC differentiation of hiPSCs. (

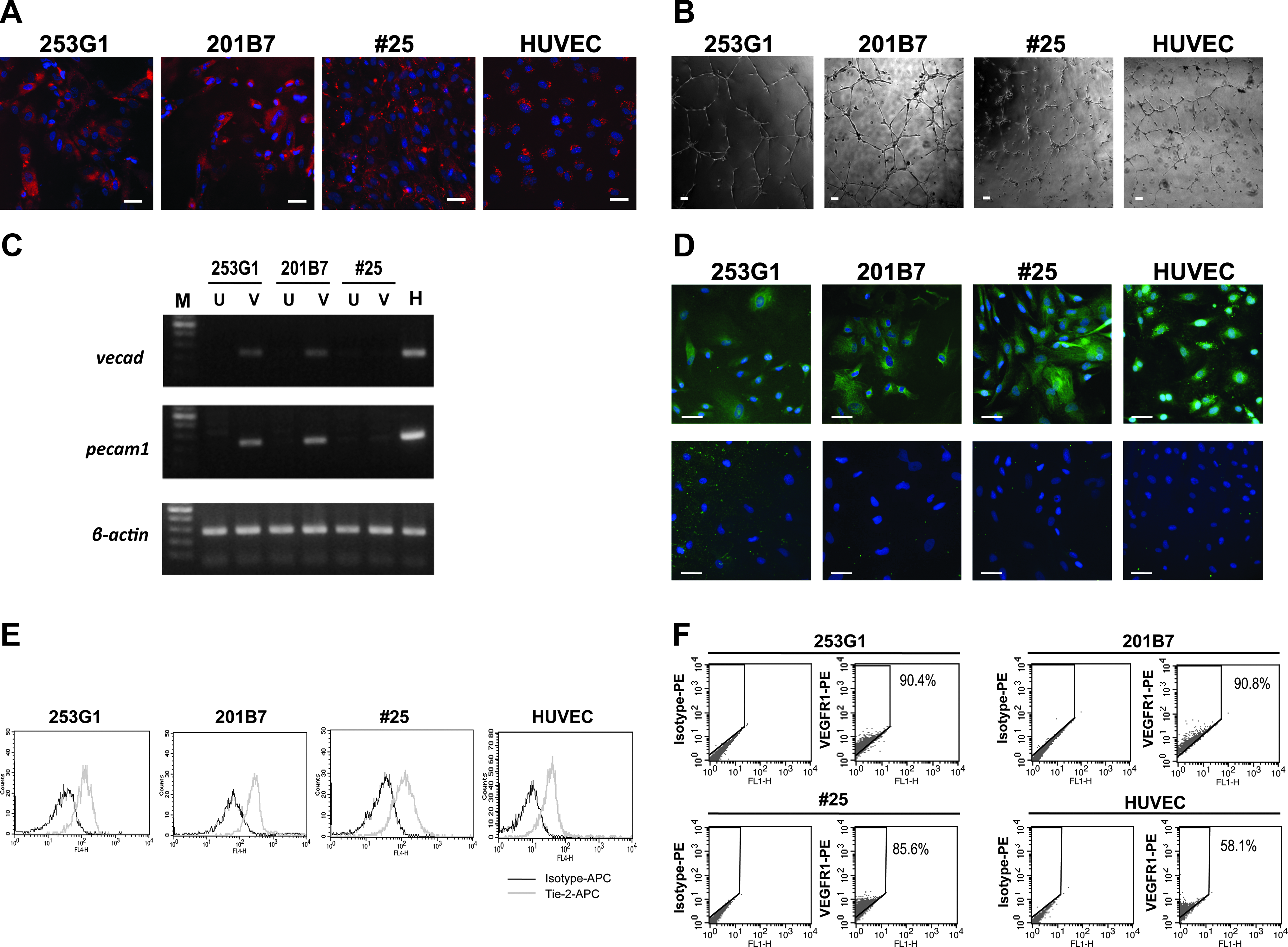
Evaluation of hiPSC-derived VECs. (
Thus, hiPSCs can generate VECs with equivalent expansion potentials to hESCs, although maturation levels of hiPSCs-derived VECs vary depending on the lines of hiPSCs.
hiPSCs can generate reproducible HPCs with comparable colony-forming activities to hESCs
We previously established a feeder-free method for the neutrophil differentiation of hESCs (Saeki et al., 2009). By our system, HPCs are generated within a unique construction named the “sac-like structure.” Within this structure, HPCs are repeatedly generated: they are reproduced within a few days after manually puncturing the sac walls and releasing the inner HPCs into the culture supernatant (Saeki et al., 2009). The reproduction process can be repeated three or four times (Saeki et al., 2009).
By applying this method, we performed neutrophil differentiation of 251G3, 201B7, and #25. All three lines successfully generated sac-like structures that were filled with abundant spheroid cells (Fig. 3A–C). However, the walls of 201B7-derived sacs seemed rather fragile because the inner spheroid cells spontaneously permeated the sac-like structures (Fig. 3B). Furthermore, we failed in reproducing spheroid cells after manually puncturing the sac walls and releasing inner spheroid cells into the culture supernatant (data not shown). In the case of 253G1, the sac walls seemed solid; however, spheroid cells were scarcely reproduced after sac wall puncturing (Fig. 3D and E). In contrast, spheroid cells were actively reproduced in the case of #25 (Fig. 3F and G). Eventually, the reproduction process persisted up to 60 days after the start of differentiation (data not shown), which was longer than that of hESCs (up to 40 days) (Saeki et al., 2009). Thus, the #25 line bears an equivalent, or even higher, spheroid cell-reproducing potential to hESCs.
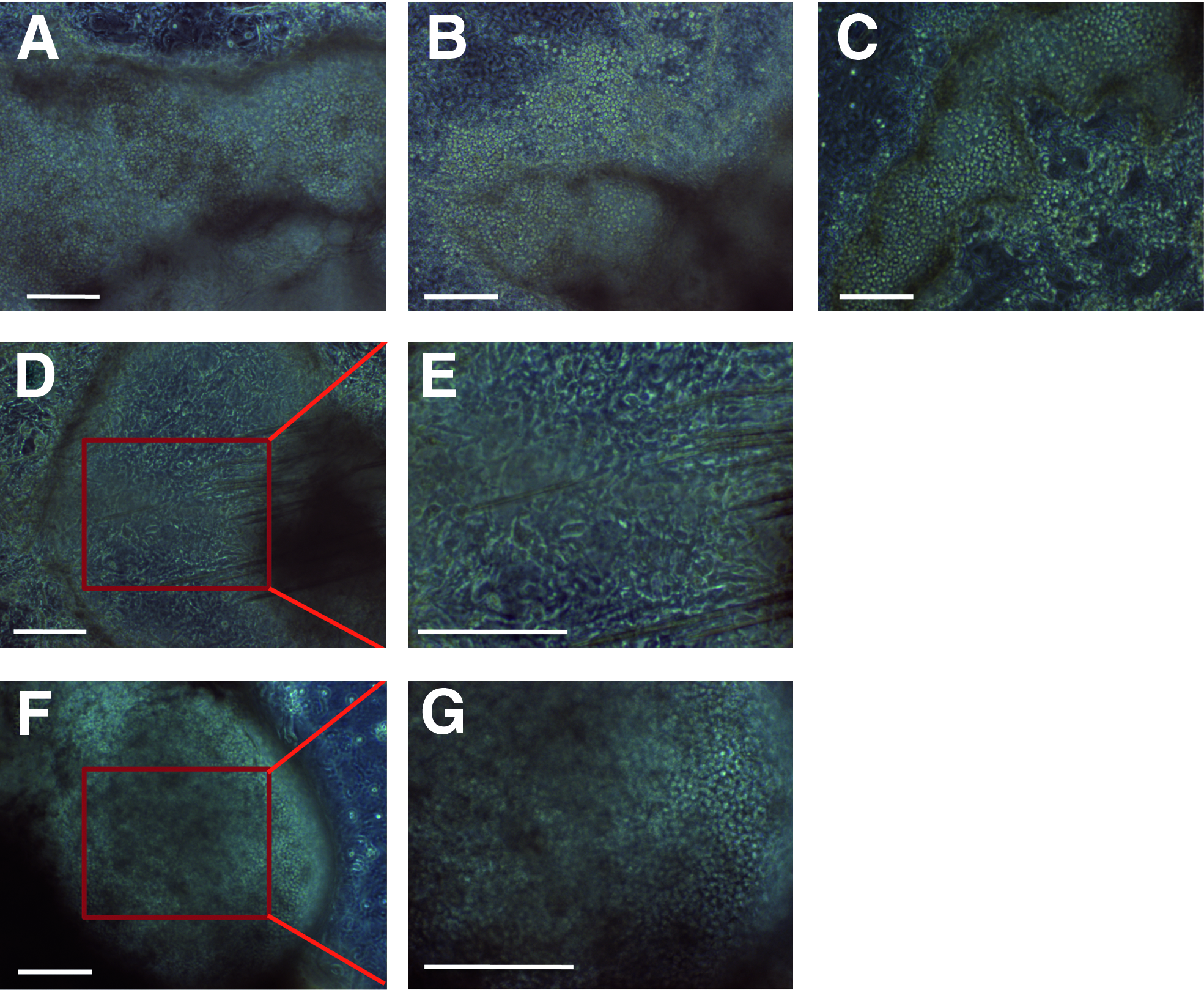
Phase contrast microscopic observation during hematopoietic differentiation of hiPSCs. (
We further evaluated the qualities of hematopoietic differentiation of 253G1, 201B7, and #25. First, cytological examinations were performed around day 30 of differentiation, when abundant neutrophil production was observed in the case of hESCs (Saeki et al., 2009). However, vast majorities of the products of 253G1 and 201B7 were macrophages (Fig. 4A, left and middle). On the other hand, various stages of granulocyte-lineage cells, from azurophilc granule-positive myeloid progenitors to segmented granulocytes, were observed in the case of #25 (Fig. 4A, right). Flow cytometric analyses demonstrated that #25-derived spheroid cells were highly positive for CD45, a pan-hematopoietic cell marker, and the majority of cells were positive for CD11b, a granulo-monocytic marker (Fig. 4B). To confirm their hematopoietic activities, colony assays were performed (Fig, 4C). The #25-derived spheroid cells demonstrated equivalent, or even higher, colony-forming unit-granulocyte (CFU-G) (8.0 ± 5.3/104 cells; n = 3), colony-forming unit-granulocyte/macrophage (CFU-GM) (12.3 ± 5.5/104 cells; n = 3), colony-forming unit-macrophage (CFU-M) (21.0 ± 3.5/104 cells; n = 3) to those of hESCs, whose average CFU-G, CFU-GM, and CFU-M per 104 cells were 2.3, 7.9, and 3.1, respectively (n = 2). Thus, hiPSCs can produce HPCs with equivalent colony-forming activities to hESCs.
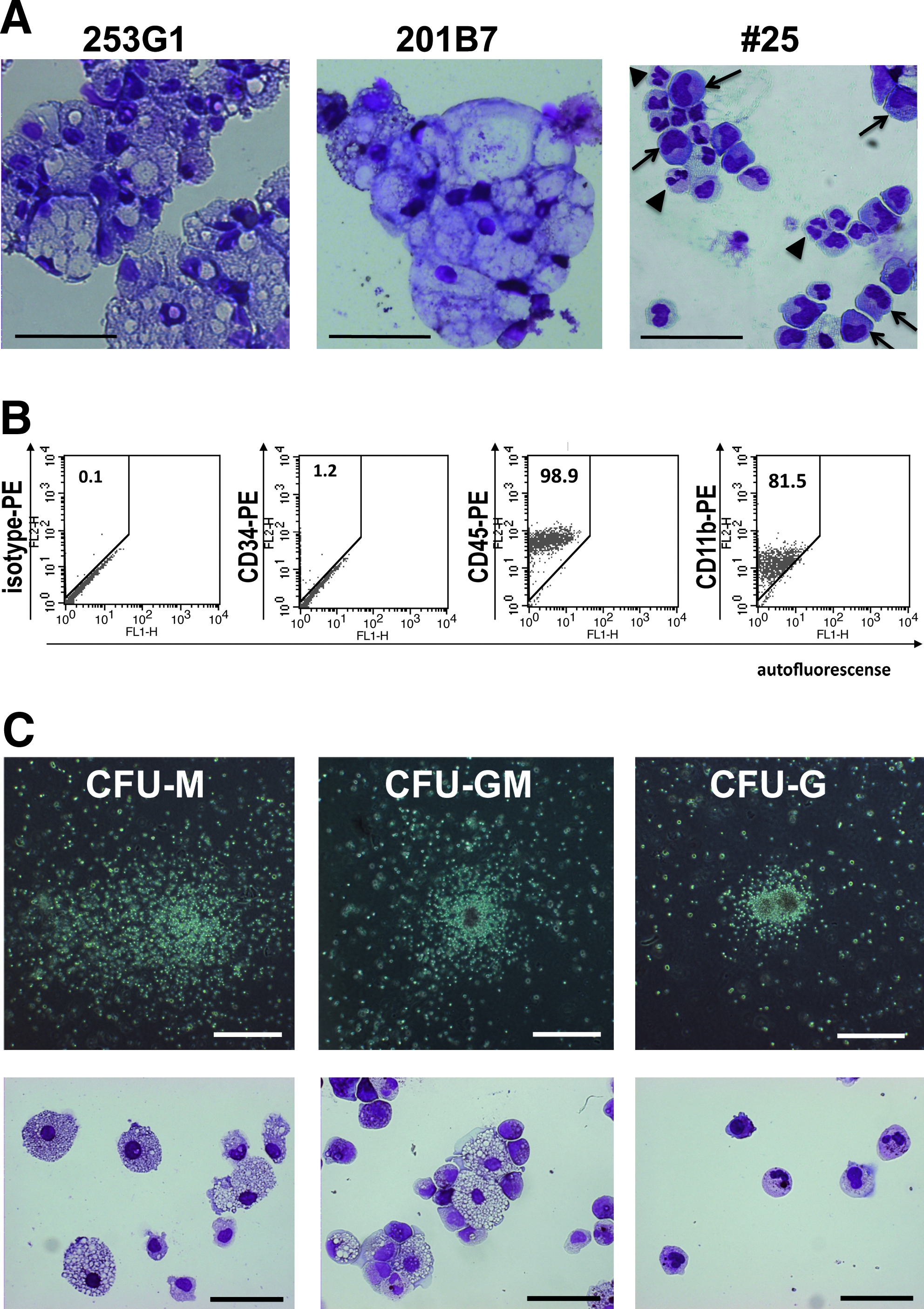
Evaluation of hematopoietic potentials of hiPSC-derived cells (
We also confirmed the functional maturation of #25-derived neutrophils by performing a superoxide production study (Fig. 5A), a phagocytosis assay (Fig. 5B), and double esterase-staining test (Fig. 5C). Interestingly, the double-esterase staining test, in which neutrophil-specific esterase is stained blue while that of nonspecific monocyte/macrophage esterase is stained brown, demonstrated that #25-derived cells showed even clearer neutrophil-specific blue staining patterns than hESC-derived ones, the majority of which showed brownish blue or bluish brown staining (Fig. 5D). Thus, hiPSCs can generate neutrophil with equivalent, or even higher, maturation levels than hESCs.
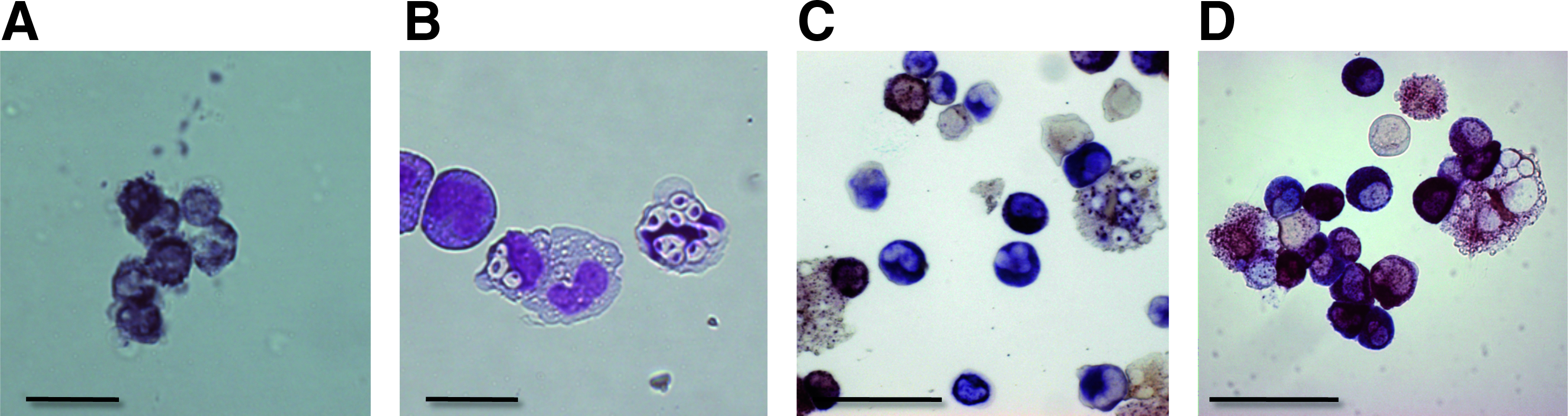
Functional assessments and special staining. (
For further assessment of possible hematopoietic potentials of 253G1-derived and 201B7-derived spheroid cells, we checked the morphologies of spheroid cells over time. Surprisingly, small round cells with high nucleus/cytoplasm ratios were detected in 253G1-derived samples around day 20 of differentiation (Fig. 6A and B), whereas no such cells were detected in 201B7-derived samples (data not shown). Interestingly, hemophagocytosis-like scenes, where the small cells were phagocytized by macrophages, were often observed (Fig. 6C). Because the morphologies of the small cells resembled to hematopoietic stem/progenitor cells, we checked their colony-forming activities (Fig. 6D). Colony assays performed around day 20 indicated that 253G1-derived cells had comparable CFU-G (3.3 ± 2.3/104 cells; n = 3), CFU-GM (3.3 ± 1.2/104 cells; n = 3), CFU-M (15.0 ± 1.0/104 cells; n = 3) to those of hESCs. On the other hand, few hematopoietic colonies were observed in the case of 201B7 at any time points (data not shown).

The hiPCS-derived differentiated cells at early phases. (
Thus, hiPSCs can generate reproducible HPCs with equivalent colony-forming activities to hESC-derived HPCs, although some lines of hiPSCs suffer from defective hematopoietic differentiation.
Discussion
In this article, we have provided the counterexamples to a previously reported finding that hiPSC-derived hemangioblast, the common progenitor of hematopoietic and endothelial cells, suffered from early senescence. In that report, hiPSC-derived HPCs was shown to have substantially decreased colony-forming activities and the majority of hiPSC-derived endothelial cells senesced after one passage (Feng et al., 2010). However, our data have clearly shown that the issue of early senescence can be overcome by selecting appropriate lines of hiPSCs and applying proper differentiation methods to them. Moreover, our results proved that retroviral insertion of reprogramming transgenes was not the cause of early senescence contrary to the discussion by the authors (Feng et al., 2010). We have also shown that, after sequential passages, hiPSC-derived VECs enter senescence as in the cases of hESC-derived VECs and primary human VECs, guaranteeing that hiPSC-derived VECs bear very low tumorigeneity, if any.
The key to our success in producing hiPSC-derived VECs that bear as high growth potentials as hESC-derived counterparts may reside, at least in part, in our usage of multiple hematopoietic cytokines in addition to VEGF. As we have shown previously, the six cytokines, SCF, IL6, IL3, BMP4, Flt3-L, and VEGF, as a whole work for the stable and high-purity production of subculturable VECs (Saeki et al, 2008). Interestingly, we are also observing that, under serum-free conditions, the presence of hematopoietic cytokine cocktail is crucial for the formation of spheres and their subsequent growth on gelatin-coated plates (M.N., unpublished finding). Thus, the usage of hematopoietic cytokine cocktail is advantageous not only for an achievement of high-efficiency differentiation but also survival and proliferation of the differentiated cells. Alternatively, the differentiation process per se, which is often followed by apoptosis, might include antiapoptotic processes as far as the differentiated cells keep surviving. In any event, stressful conditions should be avoided as much as possible from the differentiation procedures of hESCs/hiPSCs as in the case of their maintenance culture, where chromosomal aberrations are reportedly induced via stressful handling of the cells (Draper et al., 2004, Mitalipova et al., 2005).
As we mentioned, two lines of hiPSCs, 253G4 and 201B2, failed in directed differentiation into VECs. The 253G4- and 201B2-derived cells showed poor cord-forming activities and lacked VEC marker expressions, although they possessed Ac-LDL-uptaking capacities and were subculturable over 10 passages (data not shown). Their disadvantageous natures concerning VEC differentiation may be resulted from the possible line dependency in differentiation propensity among hiPSCs as reported in the case of hESCs (Osafune et al., 2008). Indeed, 253G4 and 201B2 showed very poor or no hemaotopoietic differentiation (data not shown). The finding that hiPSC lines with poor VEC-differentiating potentials bear little hemaotocyte-producing capacities seems very reasonable, because hematopoietic cells are derived from a specific population of vascular endothelial cells (Eilken et al., 2009).
Our findings together indicate that, although hiPSCs may be imposed line-dependent limitations in their differentiation capacities, they are not put inevitable fates of differentiation-dependent early senescence.
Footnotes
Acknowledgments
The authors greatly thank Professor Yamanaka at Center for iPS Cell Research and Application, Kyoto University, Japan, for generously providing the hiPSC lines (201B2, 201B7, 253G1, and 253G4). This work was supported by a Grant-in-Aid Scientific Research from the Ministry of Health, Labour and Welfare (KHD1029).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.