
Abstract
Abstract
Recent studies suggest that cellular therapies that utilize mesenchymal stem cells (MSCs), especially ones that have been neurally induced (NI), may provide a functional benefit in a wide range of neurological disorders. Recently, we developed a new method for the efficient generation of neural cells from human bone marrow (BM)-derived MSCs (hMSC). Neural induction was achieved by exposing cells simultaneously to chromatin-modifying agents and neural-inducing factors. When transplanted into injured spinal cords, these NI-hMSCs survived, differentiated, promoted tissue preservation, and significantly improved locomotor recovery of injured animals. In the current study, we sought to determine whether this methodological approach would be equally effective in generating neural-like cells from feline BM-derived MSCs (fMSC). Our long-term goal is to develop an autologous source of neural stem cells that can be used in cellular replacement therapies in large animal (feline) models of neurological disorders. Our results showed that fMSCs exhibited a neural morphology after 48–72 h of neural induction. Immunocytochemistry, ELISA, Western blot, and real-time RT-PCR studies revealed a higher level of expression of several pluripotent and neural genes in NI-fMSCs, the majority of which were expressed in untreated fMSCs at relatively low levels. We concluded that the expression of pluripotency- and neural-associated genes in unmodified fMSCs make them more pliable for reprogramming into a neural fate by manipulation with chromatin modifying agents and neural inducing factors.
Introduction
Several recent discoveries demonstrated that desirable multipotent/progenitor stem cells can either be generated by dedifferentiation of adult somatic cells into a pluripotent state, followed by redifferentiation into particular type of stem cells; or by transdifferentiation of one type of adult stem cell into another (Kim et al., 2009; Kunisato et al., 2010; Lyssiotis et al., 2007; Masip et al., 2010; Ohnuki et al., 2009; Takahashi and Yamanaka, 2006; Warren et al., 2010; Zhu et al., 2010). These findings raise the possibility that desirable tissue-specific stem cells can be generated from a patient's own somatic cells. The individual patient could serve as the source of his or her cell replacement therapy for a disease or injury condition, once a sufficient amount of condition-appropriate cells have been produced on a large enough scale.
However, most of the technology that would be required to efficiently produce an autologous supply of pluripotent or multipotent cells is still in the early stages of development. Thus, it is imperative to develop safe, easy, and efficient procedures for the generation of the desired cell phenotypes via cellular reprogramming.
Small molecules, particularly those that are involved in the regulation of chromatin and DNA covalent modifications, have been shown to be useful biochemical tools in manipulating cell stemness, commitment, and differentiation (Lyssiotis et al., 2007; Ruau et al., 2008; Schmittwolf et al., 2005; Zhu et al., 2010). Small molecules with such specific regulatory activity are starting to play a vital role in elucidating the fundamental biology of stem cells and facilitating the development of regenerative medicine therapeutic approaches.
Recently, using this relatively safer approach, we have been able to generate neural-like cells from human BM-derived MSCs (hMSC) (Alexanian, 2010b). Neural induction was achieved by exposing cells simultaneously to inhibitors of histone deacetylation, DNA methylation, and pharmacological agents that elevate cAMP levels. This new methodological approach of neural induction has been developed on the basis of our previous cell plasticity studies (Alexanian, 2005, 2007; Alexanian et al., 2008, 2010). In this approach, we used a combination strategy of reactivating the pluripotency-associated genes in the MSCs while simultaneously exposing them to neural inducing factors.
The expression of both the reactivated pluripotency markers and the newly induced neural markers in the neurally induced hMSCs (NI-hMSCs) was confirmed with immunocytochemistry, Western blot, and real-time RT-PCR techniques. ELISA studies showed that these NI-hMSCs were capable of expressing and releasing neurotrophic factors such as GDNF and BDNF.
Transplanted NI-hMSCs survived, differentiated, reduced the volume of cavity, and promoted white matter sparing in injured spinal cords (ISC) of rats and significantly improved locomotor recovery of injured animals compared to control rats that were transplanted with nonmodified hMSCs and vehicle.
Thus, hMSCs neurally modified by this original method may provide an alternative source of autologous adult stem cells that could be useful for central nervous system (CNS) regeneration via replacement of injured or diseased cells and/or providing support to CNS tissue.
The goal of this study was to test the effectiveness of this methodological approach to generate neural cells from feline BM-derived MSCs (fMSC) that can be used as an autologous source of neural stem cells in large animal (feline) models of neurological disorders.
Material and Methods
Isolation of mesenchymal stem cells from feline marrow
Feline bone marrow was harvested with a needle through the tip of the greater trochanter into the medullary canal of the cat femur and collected into 1–5 volumes MEM Alpha Medium (GIBCO, Grand Island, NY). After cells were pelleted by 500 G centrifugation, they were rinsed in the same medium. The resulting pellets were resuspended in MEM Alpha Medium containing 10% fetal bovine serum (FBS) that screened for its ability to support mesenchymal stem cell proliferation (Stem Cell Technologies, Vancouver, Canada), 100 U/mL penicillin,100 mg/mL streptomycin, and 2 mM Glutamine (Sigma, St. Louis, MO). The cells were plated at density of 1×109/cm2 in 75 cm2 plastic flasks and incubated at 37°C in a humidified atmosphere with 5% CO2. Hematopoietic and nonadherent cells were removed by a change of medium after 48 h. Expanded cells were used for neural induction studies.
Neural induction
Neural induction was performed by the method described recently (Alexanian, 2010a). Briefly, fMSCs were exposed to 200 nM trichostatin A (TSA) (inhibitor of histone deacetylases), 3 μM RG-108 (DNA methyltransferase inhibitor), 300 μM 8-BrcAMP (highly stable, biologically active form of cAMP) and 1 μM Rolipram (inhibitor of phosphodiesterases), in the medium of NeuroCult/N2 supplemented with 20 ng bFGF.
Immunocytochemistry
For immunocytochemistry, fMSCs and 24, 48, 72 h treated NI-fMSCs were fixed with 4% paraformaldehyde and stained for several immature and mature neural markers, such as A2B5, NCAM, B3T, MAP2, NeuN, neurofilament 200 (NF), Nurr1 (early marker for dopaminergic neurons), tyrosine hydroxylase (TH) (dopaminergic marker), choline acetyltransferase (ChAT) (cholinergic marker), GABAergic (GABA), and serotonin (5-HT) (serotonergic neuronal marker). First, cells were permeabilized 10 min with 0.2% Triton X-100 in phosphate-buffered saline (PBS), followed by blocking with 5% goat serum in PBS for 30 min, then incubated for 1 h with one of the following primary antibodies in PBS. Mouse monoclonal anti-A2B5 (1:200, Chemicon, Temecula, CA), rabbit polyclonal ant-NCAM (1:500, Millipore, Bedford, MA), mouse monoclonal anti-β-III-tubulin (B3T) (1:750,; Covance, Richmond, CA), rabbit polyclonal anti-MAP2 (1:500, Millipore), mouse monoclonal anti-NeuN (1:200, Millipore), rabbit polyclonal antineurofilament 200 (1:1000, Millipore), rabbit polyclonal anti-Nurr1/NOT1 (1:300, Millipore), rabbit polyclonal antityrosine hydroxylase (1:150, Millipore), rabbit polyclonal anti-ChAT (1:500, Millipore), mouse monoclonal anti-GABA (1:200, Millipore), mouse monoclonal antiserotonin (5HT, 1:50, Abcam, Cambridge, MA). Immunoreactive cells were visualized with Texas Red (TxR)-conjugated goat antirabbit IgG or fluorescent-conjugated (FITC) goat antimouse IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). To reduce fluorescence quenching, glass coverslips were mounted in ProLong Antifade reagent (Molecular Probes, Eugene, OR), and dried on microscope slides. Representative images were captured by a Nikon inverted microscope equipped with color digital camera (Spot II). Metamorph software (Universal Imaging, Ypsilanti, MI) was for analyzing the images and counting the cells. In the results section, values represent the average from two different experiments with standard deviations.
Western blot analysis
Cells grown in six-well tissue culture plates were used for preparation of whole-cell extract. After removing the media, cells were rinsed twice with PBS and incubated in 1 mL/well trypsin/EDTA (Invitrogen, Carlsbad, CA) for 5 min at 37°C. Cold, sterile 1×PBS (1 mL/well) was added to each well after incubation. The suspension was centrifuged for 5 min at 500×g. After cells were resuspended in 60–120 μL lysis buffer (50 mM Tris HCl, 1 mM EGTA, 1% SDS, 1 mM EDTA, 5 μL/mL PMSF, 1% IGPAL, 10 μL/mL protease inhibitor cocktail) and kept on ice for 30 min. The mixture was centrifuged at 16,000×g for 15 min at 4°C and the supernatants were collected. The protein concentration was determined by BCA Protein Assay Kit (Thermo Scientific, Inc., Pittsburgh, PA). The samples were frozen at −80°C. For SDS-polyacrylamide gel electrophoresis equal amounts of protein extracted from cells (10 μg) were resolved on 4–15% polyacrylamide gradient (Bio-Red, Richmond, CA). For Western blotting, proteins were transferred to nitrocellulose membrane by Trans-Blot SD Electrophoretic Transfer Cell. After blocking with 5% dry milk reconstituted in Tris-buffered saline, blots were incubated with the following primary antibodies: rabbit polyclonal anti-Sox2 (1:1000, Chemicon International), chichen polyclonal anti-c-myc (1:6000, Millipore), rabbit polyclonal anti-Nanog (1:2000, Millipore), mouse monoclonal anti-B3T (1:3000, Covance), mouse monoclonal anti-NeuN (1:1000, Millipore), mouse monoclonal anti-neuron specific enolase (NSE) (1:1000, Millipore), rabbit polyclonal anti-Nurr1/NOT(1:1000, Millipore), rabbit polyclonal anti-TH(1:800, Millipore), rabbit polyclonal anti-ChAT (1:1500, Millipore), mouse monoclonal anti-β-Actin (1:7000, Millipore). After incubation with goat anti-rabbit/mouse HRP-conjugated secondary antibody (1:6000, Pierce Chemical Company, Rockford, IL), or rabbit antichicken HRP conjugated secondary antibody (1:6000, Thermo Scientific), the protein bands were detected using the chemiluminescent substrate SuperSignal West Dura or SuperSignal West Maximum Sensitivity Substrate (Pierce Chemical Company) and by capturing and digitizing the images with Kodak Image Station 2000MMT. The experiments were carried out in duplicates and the data is shown as standard error of the mean. The statistical significance was assessed by one-way analysis of variance (AVOVA) followed by Tukey's pair comparisons. Values of p<0.05 were considered significant.
Quantitative real-time RT-PCR analysis
Total cellular RNA was extracted from untreated fMSCs and from 24-, 48-, and 72-h treated NI-fMSCs by using the PureZOL RNA Isolation Reagent and the Aurum Total RNA Mini Kit (Bio-Rad). A cDNA was synthesized from 1 μg of total RNA using reverse transcriptase (iScript cDNA Synthesis Kit, Bio-Rad). For quantitative RT-PCR, real-time PCR was conducted on a Bio-Rad iCycler. Genes amplified by 0.5 μM of both sense and antisense primers (Table 1) and amplification was monitored using iQ SYBR Green Supermix (Bio-Rad).
To check whether amplification yields PCR products with a single molecular weight, the PCR products were electrophoresed on 1% agarose gels containing ethidium bromide. To eliminate the possibility of nonspecific amplification or primer–dimer formation a melting curve analysis was performed after the amplification phase. Relative gene expression was analyzed by 2−ΔΔCT method. The experiments were carried out in triplicate and repeated twice. The statistical significance was assessed by one-way AVOVA followed by Tukey's pair comparisons. Vvalues of p<0.05 were considered significant.
Emax immunoassay system
To study the ability of NI-fMSCs to release neurotrophic factors that are important for neuronal survival and growth, the Emax immunoassay system for NGF BDNF, NT3, and GDNF was used. The immunoassay was conducted according to the product protocol (Promega, Madison, WI). Flat-bottom 96-well plates were coated with a Polyclonal Antibody (pAb) to one of the neurotrophic factors which binds soluble GDNF, BDNF, NT-3, and NGF. Samples (100 μL) from treated 72-h fMSCs that were grown for an additional 24 h prior to immunoassay in defined medium (Neurobasal A/B27) were added in antibody-coated wells. The captured neurotrophic factors were bound by secondary specific monoclonal antibodies (mAbs). The samples were then washed and the amount of specifically bounded mAbs was detected using a species-specific antibody conjugated to horseradish peroxidase (HRP). The unbound conjugates were removed by washing and the samples were incubated with a chromogenic substrate. After 30 min, the reactions were stopped and the color change was measured at 450 nm on a plate reader. The NGF, BDNF, NT-3, and GDNF standards provided with this system were used for generation of a linear standard curve from 7.8–500 pg/mL. Data represent the means of two independent experiments performed in duplicate.
Results and Discussion
FMSCs grown in MEM Alpha Medium supplemented with 10% fetal bovine serum, exhibited a spindle-shaped and flattened morphology. Morphologically, fMSCs appeared very similar to their rodent and human counterparts.
Immunocytochemical results showed that 10–20% of untreated fMSCs exhibited low immunoreactivity to neural markers such as B3T, NCAM, A2B5, MAP2, NeuN, NF, Nurr1, TH, and ChAT. The percentage of cells that were highly positive for B3T, NCAM, A2B5, MAP2, NeuN, and NF was 3–7%, and for Nur1, TH, and ChAT was less than 0.5–1% (Fig. 1a–f). The percentage of cells that were positive to B3T, A2B2, NCAM, MAP2, NeuN, and NF was gradually increased during the next 2 days during the treatment, and after 72 h they were 95±11.02, 85.64%±7.85, 87.58%±9.23, 85.45%±8.45, 82.03%, and 77.54%±8.05, respectively (Fig. 1g–l). The percentage of cells positive for dopaminergic marker Nurr1 and TH, and cholinergic marker ChAT were 58.42%±7.51, 50.02%±8.34, and 62.34±8.45, respectively (Fig. 1m–r). No cells positive for GABA and 5-TH were observed. In total, more than 95% of cells were positive to neural markers. Only a small percentage (less than 5%) of cells still retained a fibroblastic morphology.
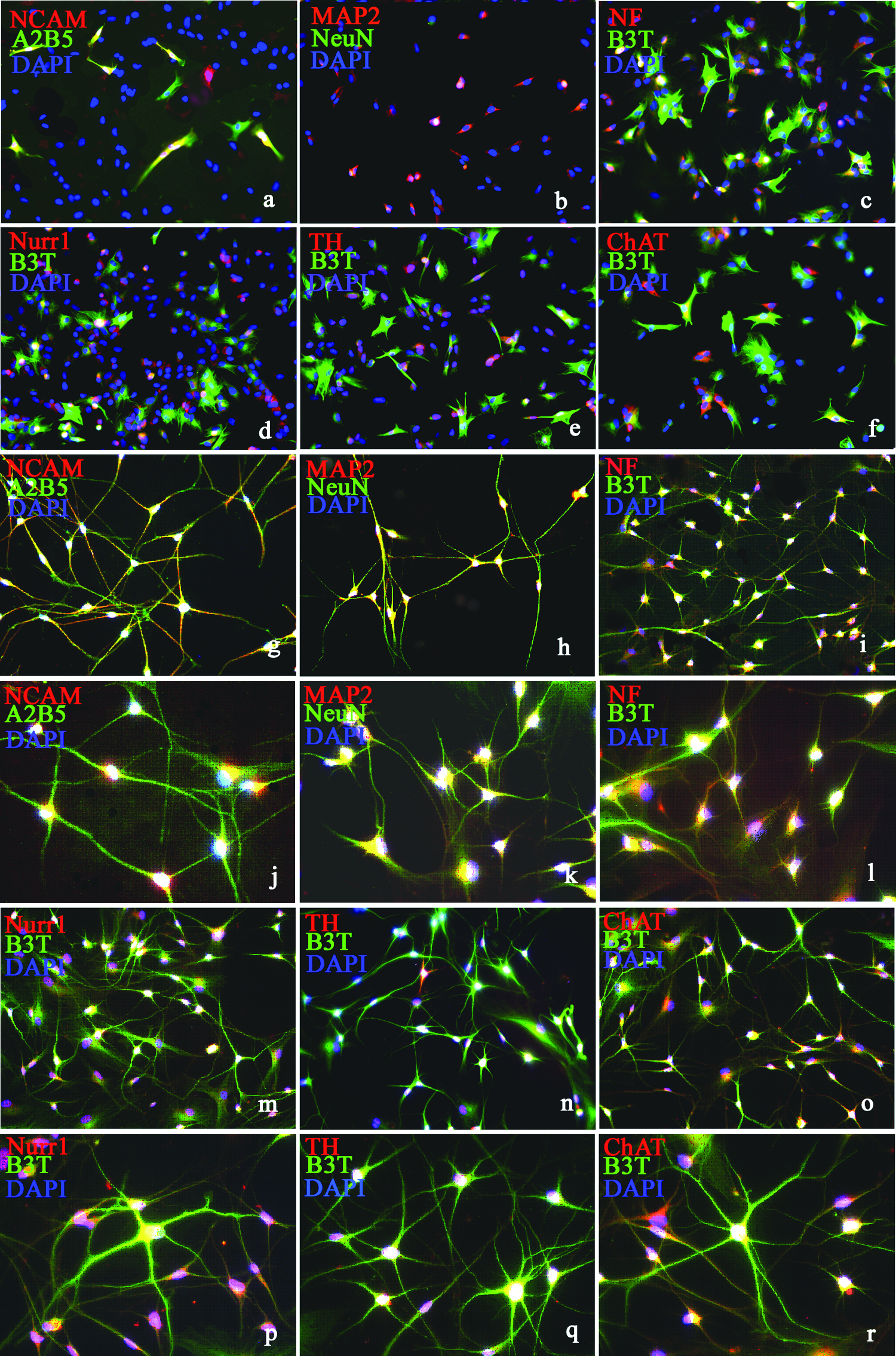
Morphological and immunocytochemical characterization of unmodified and NI-fMSCs. Expression of neural markers B3T, NCAM, A2B5, MAP2, NeuN, NF, Nurr1, TH, and ChAT in unmodified fMSCs (
ELISA studies demonstrated that NI-fMSCs released neurotrophic factors GDNF (144.4±7.51 pg/mL/1×105/day) and NGF (377.1±90.11 pg/mL/1×105/day).
Gene expression studies by real-time RT-PCR showed that native fMSCs expressed several pluripotent markers such as Oct-4, Nanog, Sox2, cMyc, and KLF4. With neural induction, expression levels of Sox2, Klf4, Nanog, and Oct4 were gradually increased during the next 3 days in culture (Fig. 2). The expression level of cMyc was highest at 24 h and then decreased during the next 2 days of the treatment but still stayed high in comparison to control (Fig. 2). The expression levels of most of the immature and mature neural markers also were gradually increased and reached to their highest level at 72 h of the treatment (Fig. 3).
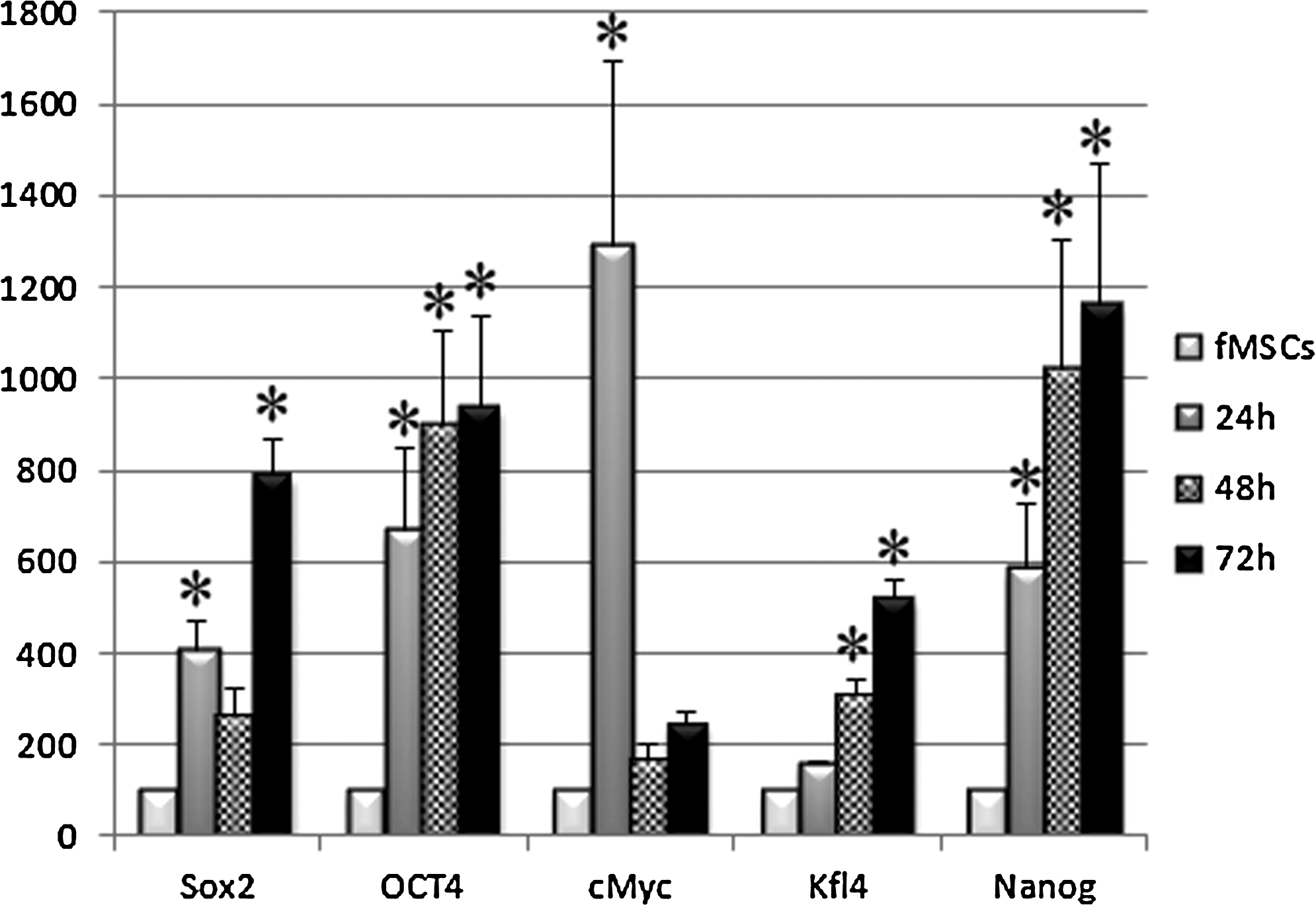
Real-time RT-PCR analysis of pluripotent gene expression in unmodified fMSCs and in NI-fMSCs after 24, 48, and 72 h of treatment. Expression levels of Sox2, Klf4, Nanog, and Oct4 were gradually increased during the next 3days and most of them were significantly different from the control after 24 h of the treatment. The expression level of cMyc was significantly increased after 24 h of the treatment but decline during the next 2 days.

Real-time RT-PCR analysis of the immature and mature neural gene expression in unmodified fMSCs and in NI-fMSCs after 24, 48, and 72 h of the treatment.
The expression of some of the pluripotent genes and most of the neural genes in unmodified and NI-fMSCs was confirmed by Western blot. The expression levels of pluripotent markers Sox-2, cMyc, and Nanog (except of Oct4 and Klf4) in NI-hMSCs were gradually increased and became significantly different from unmodified hMSCs after 48 h and 72 h of the treatment (Fig. 4). The lack of the expression of Oct-4 and Klf4 suggests that either the expression level of these proteins was very low or they were expressed only at the mRNA level. Western blot studies also revealed a gradual increase of neuroectoderamal gene expression in NI-fMSCs, which became significantly different from the control after 48–72 h of the treatment (Fig. 5). This increase in neural genes expression was accompanied with significant morphological changes of fMSCs into neural phenotypes. Interestingly, neural transformation rate of fMSCs was much faster (only 3 days) in contrast to mouse and human MSCs, which usually took about 2 weeks.

Western blot analysis of pluripotent gene expression in unmodified fMSCs and in NI-fMSCs after 24, 48, and 72 h of the treatment. Expression of genes normalized to β-actin (image on the left). The expression levels of Sox2, Nanog, and cMyc in treated cultures were significantly different from untreated fMSCs after 48 or 72 h of the treatment.

Western blot analysis of neural genes expression in unmodified fMSCs and in NI-fMSCs after 24, 48, and 72 h of treatment. Expression of genes normalized to β-actin (image of the left). The expression levels of neural genes in NI-hMSCs were significantly higher from untreated hMSCs after 48 and/or 74 h of the treatment.
Thus, fMSCs manipulated with the neural induction protocol used in this study, turned into neural-like cells more easily and efficiently compared to mice and human MSCs.
We concluded that the higher plasticity of fMSCs could be explained by the moderate expression of several pluripotent and neural genes in unmodified fMSCs.
Footnotes
Acknowledgments
This work was supported by VA Medical Research and the Department of Neurosurgery at the Medical College of Wisconsin. The authors thank Christy Stadig and Kyle Stehlik for technical assistance.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.