
Abstract
Abstract
Manipulation of mammalian embryos and gametes in vitro reduces viability. Specific causes for these reductions are still largely undetermined. Accumulating evidence suggests that survival rates and developmental competency may be reduced following disruptions in the epigenetic regulation of gene expression. Chromatin-based epigenetics can regulate the transcriptome through the establishment of different transcriptionally permissive and repressive chromatin environments. Recently, support has been gathering for the hypothesis that the in vitro embryo displays reduced viability due to abnormal remodelling of the paternal chromatin, which is hypothesized to result in global transcriptional repression. In this study, we have used quantitative real-time PCR to document the effect of in vitro culture on the transcription of genes that code for proteins that are directly involved in the establishment of chromatin environments. We compare in vitro embryos to embryos generated through parthenogenetic activation to determine how the absence of paternal chromatin remodeling affects transcriptional activity. Through these studies, we show that the expression of many genes encoding for histone proteins and other modifiers involved in chromatin-based epigenetic regulation are perturbed by in vitro culture. In addition, we show that the expression of many candidate genes was reduced in in vitro embryos but not in parthenogenetic embryos. These results support the hypothesis that events linked to remodeling of paternal chromatin may influence transcriptional activity in the in vitro embryo and that chromatin-based reprogramming events in developing embryos are dynamically responsive to prevailing conditions.
Introduction
This study aimed to test the hypothesis that different developmental milieu affects the expression of genes coding for histone proteins and chromatin remodelers in mouse preimplantation embryos. A candidate qRT-PCR approach was used to document the transcript levels of selected genes of interest in pools of three embryos. We have specifically chosen to use a candidate qRT-PCR approach over a microarray approach to avoid the large pooling and amplification of embryo samples that have been shown to limit sensitivity and introduce bias into transcriptome based experiments (Bower et al., 2007; Gardner et al., 2004).
To test the different developmental milieu hypothesis, we have compared expression levels of candidate genes in in vivo developing embryos with those of embryos developing in vitro. Previous research has suggested that the aberrant expression of genes in vitro embryo may be caused by the deregulation of paternal chromatin. This is hypothesized to be initiated in some as yet unknown way by the in vitro culture environment and which inadvertently results in reduced transcriptional activity (Burton and Torres-Padilla, 2010; Ma et al. 2001; Mayer et al., 2000; Young and Beaujean, 2004). To evaluate whether the remodeling of paternal chromatin may influence gene expression we have also assayed the expression of candidate genes of interest in parthenogenetic embryos, which develop in the absence of paternal chromatin integration and remodeling.
The model we adopt compares transcript levels of candidate genes of key chromatin proteins. These candidate genes included 11 mouse histones [polyadenylated Hist2h2be (H2b), polyadenylated Hist2hac (H2a), H2afx, H2afz, H2afy, H2Afy2, H2afb2 (H2A.Bbd), Cenpa, Hist1h3c (H3.1), Hist2h3c (H3.2), H3f3b (H3.3b)] three specific histone remodelers [Smarcb1 (a subunit of Swi/Snf), Hira, Chafia (Caf-1)], and four other histone-related factors [Hplbp3 (Hp1), Hdac1, Dnmt3b, and Dnmt3l)]. Transcript levels for each of these genes of interest were assayed at the zygote, two-cell, morula, and blastocyst stages of mouse preimplantation development.
Methods
Ethics
The Animal Ethics and Experimentation Committees of the University of Queensland approved all experiments on mice. These committees are approved by the National Health and Medical Research Council of Australia.
Sample collection
For this study, expression and protein levels of candidate genes in embryos developing in vitro were compared to in vivo developing counterparts. For these comparisons, the variable is the developmental milieu of the embryos. All other aspects of the embryos are identical, including heritage, superovulation, mating times, and collection points, which was in excess of 75%.
The collection of all embryos was performed in KSOM–HEPES media (Lawitts and Biggers, 1993) incubated in a humidified atmosphere of 5% O2, 5% CO2, and 90% N2 at 37°C using a MINC incubator (William A. Cook Australia PTY. LTD, Brisbane, QLD, Australia). The culture of all embryos was performed in KSOM media, warmed to 37°C, and preequilibrated in a humidified atmosphere of 5% O2, 5% CO2, and 90% N2. All embryos were collected from female 8–10-week-old Quackenbush mice, superovulated by interperitoneal injection of 10 IU PMSG (Folligon, Intervet, www.intervet.com.au/) and 10 IU hCG (Chorulon, Intervet) 48 h apart.
For in vivo and in vitro developing embryo collections, superovulated females were paired with males and left overnight to mate. For in vivo developing embryo collection, embryos from mated females were collected at the zygote, two-cell, morula, and blastocyst stages (20-, 44-, 68-, and 92-h post-hCG, respectively) into KSOM–HEPES media. Cumulus cells were removed with 0.05 mg/mL hyaluronidase (Sigma, St. Louis, MO) in KSOM–HEPES, embryos washed twice in fresh KSOM–HEPES media, washed a further two times in phosphate-buffered saline (PBS) and snap frozen in a minimal amount of PBS in pools of 3 at −80°C.
For in vitro embryos, zygotes were collected from successfully mated females at 16-h post-hCG and cumulus cells were removed from zygotes with 0.05 mg/mL hyaluronidase (Sigma; www.sigma.com) in KSOM handling media followed by washing in fresh KSOM–HEPES media. Embryos were then washed twice in drops of preequilibrated KSOM and cultured in fresh KSOM at a density of one embryo/μL media. Embryos were removed from culture at 20-, 44-, 68-, and 92-h post-hCG, washed twice in PBS, and frozen in a minimal amount of PBS in pools of 3 at −80°C.
For parthenogenetic embryo production, females were superovulated with PMSG and hCG as described above; however, these mice were not mates but were placed overnight into a room containing sexually mature male mice for pheromone stimulation. Oocytes were harvested at 12-h post-hCG, denuded of cumulus cells by treatment with 0.05 mg/mL hyaluronidase in KSOM–HEPES media, and then activated by treatment with 10 mM strontium chloride in KSOM media for 2 h. Activation treatment by 10 mM strontium chloride was found to be the best activation procedure of three tested procedures, achieving morphologically healthy blastocysts at formation rates above 75% (Supplementary Fig. S1; http://www.liebertonline.com.cell). Following activation, all embryos were washed twice in drops of preequilibrated KSOM under paraffin oil and cultured for a further 2 h in preequilibrated KSOM with 2 μg Cytochalasin D (Sigma). Embryos were washed twice in drops of preequilibrated KSOM under paraffin oil and cultured in KSOM at a density of one embryo/μL. To collect zygotes, two-cell, morulae, and blastocysts embryos were removed from culture at 20-, 44-, 68-, and 92-h post-hCG washed twice in PBS and frozen in a minimal amount of PBS in pools of 3 at −80°C.
Collection times (20-, 44-, 68-, and 92-h post-hCG) were strictly adhered to for all embryo collection. In addition to this, only embryos that appeared healthy and aligned with the characteristic morphology of each desired stage were selected.
Nucleic acid extraction and synthesis of complementary DNA (cDNA)
Nucleic acids were extracted using a Dynal® mRNA micropurification kit (Invitrogen, San Diego, CA; www.invitrogen.com). Extracted mRNA was immediately reverse transcribed using SuperScript™ III (Invitrogen) as per manufacturer's recommendations. For embryos, the entire mRNA sample was reverse transcribed in a final volume of 30 μL. The cDNA was purified with QIAquick PCR purification columns (Qiagen, Chatsworth, CA; www.qiagen.com) as per manufacturer's guidelines except for the elution step, where embryo cDNA was resuspended in 120 μL of elution buffer. A total of 0.05 embryo equivalent cDNA was used in each reaction. Embryo equivalents were determined using the Qubit Quantitation Platform (Invitrogen).
Quantitative real-time PCR (qRT-PCR)
Eleven mouse histone variants [polyadenylated Hist2h2be (H2b), polyadenylated Hist2hac (H2a), H2afx, H2afz, H2afy, H2Afy2, H2afb2 (H2A.Bbd), Cenpa, Hist1h3c (H3.1), Hist2h3c (H3.2), H3f3b (H3.3b)], three specific histone remodelers [Smarcb1 (a subunit of Swi/Snf), Hira, Chafia (Caf-1)], and four other histone-related factors [Hplbp3 (Hp1), Hdac1, Dnmt3b, and Dnmt3l] were assayed (for primer specifics refer to Supplementary Table S1). Gene expression was quantified using SYBR Green qRT-PCR on an ABI PRISM 7900HT sequence detector (Applied Biosystems, Bedford, MA; www.appliedbiosystems.com) using cycling parameters defined by the manufacturer. To ensure the absence of contamination, no-template controls were included in all assays.
All known mouse genes for each histone were aligned using the Australian National Genomic Information Service (ANGIS). These alignments were used to ensure that primers were designed outside of shared histone domains and thus were unique to the transcript of interest. Primers were designed using the Primer3 interface (v. 0.4.0, http://frodo.wi.mit.edu/). To ensure the absence of primer dimers, the melt curves of each primer pair were assessed. The identity of PCR products was validated by direct cycle sequencing with amplification primers using ABI BigDye terminator chemistry 3.1 (Applied Biosystems) on an ABI 3130xl Genetic Analyzer system (Applied Biosystems). All expression normalization calculations were based on the Pfaffl mathematical model (Pfaffl, 2001) and take into consideration varying PCR efficiencies. For each PCR primer pair PCR efficiencies were calculated using LinRegPCR software version 7.0 (Ramakers et al., 2003). Transcripts that were not detectable (where qRT-PCR resulted in Ct values >31) are graphically represented by “ND.”
Selection of an appropriate endogenous reference gene for preimplantation qRT-PCR
Prior to undertaking qRT-PCR to assay candidate gene expression, gene stability assays were conducted to determine which of seven commonly used endogenous referencing genes was the most stably expressed in preimplantation mouse embryos. To do this, the expression of seven selected endogenous reference genes was assayed, including conserved helix-loop-helix ubiquitous kinase (Chuk), glyceraldehyde 3-phosphate dehydrogenase (Gapdh), hypoxanthine phosphoribosyltransferase 1 (Hprt1), peptidylprolyl isomerize A (Ppia), TATA box binding protein (Tbp), tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide (Yhwaz), and ribosomal protein, large, P0 (Rplp0), (for primer pairs, see Supplementary Table S2). The expression of these potential endogenous reference genes were evaluated across all stages of preimplantation development (including the zygote, two-cell, morula, blastocyst, and hatching blastocyst stages at 20-, 44-, 68-, 92-, and 116-h post-hCG, respectively) using geNorm software (http://medgen.ugent.be/∼jvdesomp/genorm/). geNorm software is able to rank genes according to their stability by measuring their “stability value” which is termed “M.” The lower the M value, the more stable a gene is across all stages or samples in which gene expression has been measured. Of the seven tested housekeeping genes, Rplp0 was found to have the lowest M value (1.43) averaged across all embryo stages assayed (Supplementary Fig. S2). The stability of two stable genes (Rplp0 and Ppia) and the two unstable genes (Gapdh and Tbp) was then tested in embryos generated by different means (in vivo, in vitro, and parthenogenesis) at different stages (including the zygote, two-cell, morula, and blastocyst stages 20-, 44-, 68-, and 92-h post-hCG, respectively). The M value for these genes in each population of embryos was calculated. Of the four tested housekeeping genes, Rplp0 was found to have the lowest M value (0.62) averaged across all embryo types and assayed stages (Supplementary Fig. S3).
To quantify and normalize gene expression levels of candidate genes in this study, Rplp0 served as an endogenous reference gene. Expression normalization for each candidate gene candidate was calculated using a comparative Ct method to determine relative quantification (Pfaffl, 2001).
Statistical analysis
Both one-way and two-way ANOVA with posttests, and in some instances Student's t-tests, were used for statistical analyses (GraphPad Software, San Diego, CA; www.graphpad.com). Gene expression levels of in vitro cultured embryos were compared to the corresponding developmental stages of in vivo embryos. For the parthenogenetic embryos, the corresponding in vitro cultured embryo stages served as the comparison.
Acid extraction of histone protein
Groups of 30 zygotes, 30 two-cell embryos, 30 morula, and 20 blastocysts were collected into siliconized Eppendorf tubes in a minimal volume of PBS (<2 μL), snap frozen on dry ice and kept at −80°C. Embryos were thawed in a triple lysis buffer (50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 0.1% SDS, 0.6 mM PMSF, 1 μg/mL of aprotinin, leupeptin, and N-tosyl-L-phenylalanine chloromethyl ketone, 1.0% Igepal CA-630, and 0.5% sodium deoxycholate), vortexed for 30 s and incubated on ice for 15 min. Five micrograms of calf thymus histone H1 (Sigma) was added to the samples (to serve as a carrier protein) followed by the addition of sulphuric acid (final concentration 0.4 N). Samples were vortexed and spun down briefly, then incubated for 1 h on ice. Samples were centrifuged at 14,000 rpm for 30 min at 4°C and the supernatant transferred to siliconized Eppendorf tubes, where 10 volumes of ice-cold acetone was added and histones left to precipitate at −20°C overnight. The following day, samples were centrifuged at 14,000 rpm for 30 min at 4°C and washed twice in 80% ice cold acetone. The histone protein pellet was allowed to dry for 1 h at 45°C before resuspension in water. Histone proteins were used for electrophoresis within a week of extraction.
Western Immunoblotting
Frozen histone isolate samples were thawed in 10 μL of 10% Laemmli sample buffer [10% (w/v) SDS, 1% (v/v) glycerol, 0.125 g bromophenol blue/L, 0.125 g xylene cyanol/L, 100 mM dithiothreitol/L, and 125 mM Tris-HCl/L (pH 6.8)] (Laemmli, 1970). Prior to electrophoresis, samples were boiled for 5 min. Electrophoresis was through a NuPAGE® Novex 4–12% Bis-Tris Gel 1.0 mm (Invitrogen) polyacrylamide at a constant voltage of 180 V for 50 min. The gel and eight sheets of blotting paper were soaked in blotting buffer (20% Methanol, 3 g/L glycine, 0.15% ethanolamine, 0.5 g/L SDS). Proteins were transferred to 0.45 μm Amersham Hybond™-C Extra nitrocellulose membranes (GE Life Sciences, Piscataway, NJ; www.gelifesciences.com) previously soaked in blotting buffer for 90 min at constant current 225 mA. Before the addition of blocking solution, total protein loading was checked using Ponceau S staining (Sigma) as per manufacturer's specifications. Membranes were then treated with blocking solution [5% bovine serum albumin (BSA), 0.1% Tween-20, in phosphate-buffered saline (PBS)] for 1 h at room temperature with gentle shaking, followed by two 10-min washes with PBS containing 0.1% Tween-20. Primary antiserum diluted in PBS containing 0.1% Tween-20, 1% BSA was applied and allowed to bind at 4°C overnight with gentle shaking followed by five 10-min washes in PBS containing 0.1% Tween-20. H2AFZ primary antiserum (ab4174) was diluted 1:1000 (AbCam, Cambridge, MA; www.abcam.com). Secondary antibody was rabbit antigoat HRP conjugated (AbCam), diluted 1:10,000 in PBS containing 0.1% Tween-20, 1% BSA, and allowed to bind for 1 h at room temperature with gentle shaking. Membranes were washed five times for 10 min each in PBS containing 0.1% Tween-20. Proteins were visualized using a Super-signal West Femto Maximum Sensitivity Detection Kit (Pierce, Rockford, IL; www.piercenet.com) according to the manufacturer's protocol. Due to many nonspecific bands when staining with the H2AFZ antibody, an immunospecificity test was also performed using a H2AFZ-specific peptide (ab11681) (AbCam). To test for specificity, twin blots were run using histone proteins extracted from 50 unfertilized oocytes. One blot was incubated with H2AFZ primary antiserum (1:1000), the second was incubated with H2AFZ primary antiserum (1:1000) and 1 μg H2AFZ peptide. Secondary antiserum binding was conducted as described above.
Results
Constituents of the chromatin based epigenetic system not expressed in mouse preimplantation embryos
Polyadenylated H2a was not detectable in the zygotes, two-cell embryos, morulae, or blastocysts that had developed in vitro or through parthenogenetic activation. In addition, transcripts for H2A variants H2A.Bbd and H2afy2 and the H3 variant transcripts H3.1 and H3.2 were not detected in any embryo of any origin (data not shown).
Expression of pericentric heterochromatin protein 1 (Hp1) was elevated in parthenogenetic embryos
Both in vitro and parthenogenetic manipulations perturbed the developmental profile of Hp1 transcripts in zygotes (two-way ANOVA, p<0.001). For in vitro zygotes, Hp1 levels were reduced by about 50% from those developing in vivo, but at subsequent stages levels matched those of embryos developing in vivo. In contrast, parthenotes embryos showed an almost fourfold elevation in Hp1 transcript levels above that of similarly treated but naturally fertilized counterparts. No differences in Hp1 transcript abundance between treatments were observed at the other developmental stages (Fig. 1).

Relative gene expression of the heterochromatin protein 1 (Hp1) in preimplantation mouse embryos. Hp1 transcript levels were assayed at the zygote, two-cell, morulae, and blastocyst stages (20, 44, 68, and 92 h, respectively). Each column represents mean normalized expression (performed using an endogenous reference gene approach, with the housekeeping gene Rplp0). mRNA was isolated from pools of three embryos (each reaction contains 0.05 embryo equivalent cDNA). Statistical tests were carried out to detect differences in gene expression levels between in vivo and in vitro embryos, and between in vitro and parthenogenetic embryos using two-way ANOVA with Bonferroni post tests. Graphs are representative of three individual experiments, error bars represent±SEM with significant differences between embryo pools at each stage represented by ***(p<0.001).
Expression of histone deacetylase 1 (Hdac1) and DNA methyl transferase 3B and 3L (Dnmt3b and Dnmt3l) genes was affected by both in vitro culture and parthenogenesis
At the zygote stage, Hdac1 transcript levels were very low and no treatment differences were observed. Differences between embryos in different treatment groups were detected at the two-cell stage and during development to morulae (two-way ANOVA, p<0.01 and p<0.001, respectively). At these stages, in vitro developing embryo levels of Hdac1 were lower than in the in vivo controls. Parthenote Hdac1 levels were elevated compared to levels within in vitro developing embryos at the two-cell and morula stages (two-way ANOVA, p<0.05 and p<0.01, respectively) (Fig. 2).
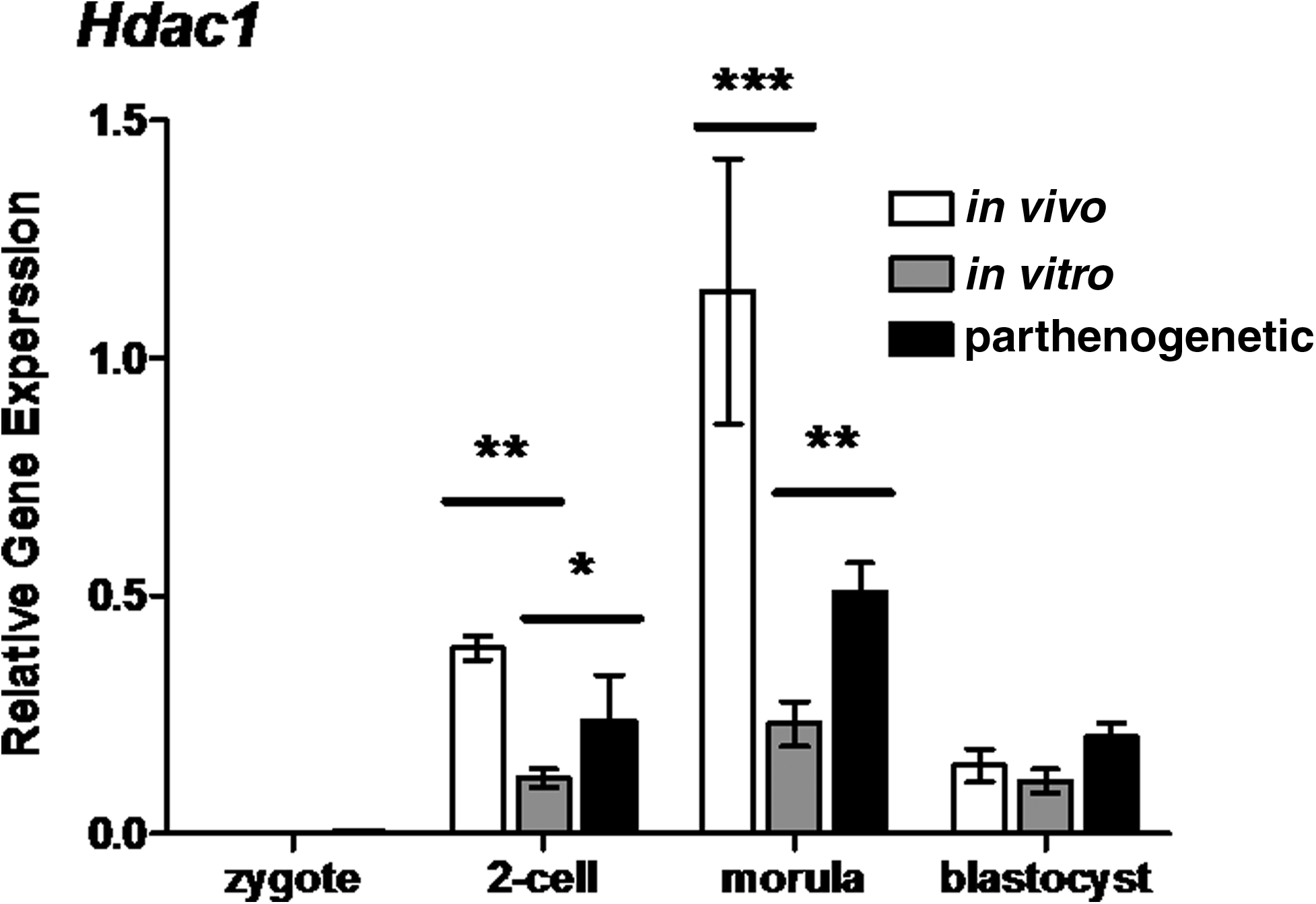
Relative gene expression of the histone deacetylase 1 (Hdac1) in preimplantation mouse embryos. Hdac1 transcript levels were assayed at the zygote, two-cell, morulae and blastocyst stages (20, 44, 68, and 92 h, respectively). Each column represents mean normalized expression (performed using an endogenous reference gene approach, with the housekeeping gene Rplp0). mRNA was isolated from pools of three embryos (each reaction contains 0.05 embryo equivalent cDNA). Statistical analysis performed between in vivo and in vitro columns and in vitro and parthenogenetic columns using two-way ANOVA with Bonferroni post tests. Graphs are representative of three individual experiments, error bars represent±SEM with significant differences between embryo populations at each stage represented by *(p<0.05), **(p<0.01), and ***(p<0.001).
DNA methyltransferase gene expression at the zygote stage was also perturbed by in vitro culture and parthenogenetic activation. The expression of Dnmt3b was not affected by in vitro culture, but parthenogenetic activation increased zygote levels (two-way ANOVA, p<0.01) (Fig. 3A). Conversely, in vitro culture did affect the expression of Dnmt3l, which was reduced in in vitro developing embryos compared to in vivo controls (two-way ANOVA, p<0.001) (Fig. 3B). Dnmt3l levels did not differ between in vitro developing embryos and parthenogenetic embryos at any assayed stage.
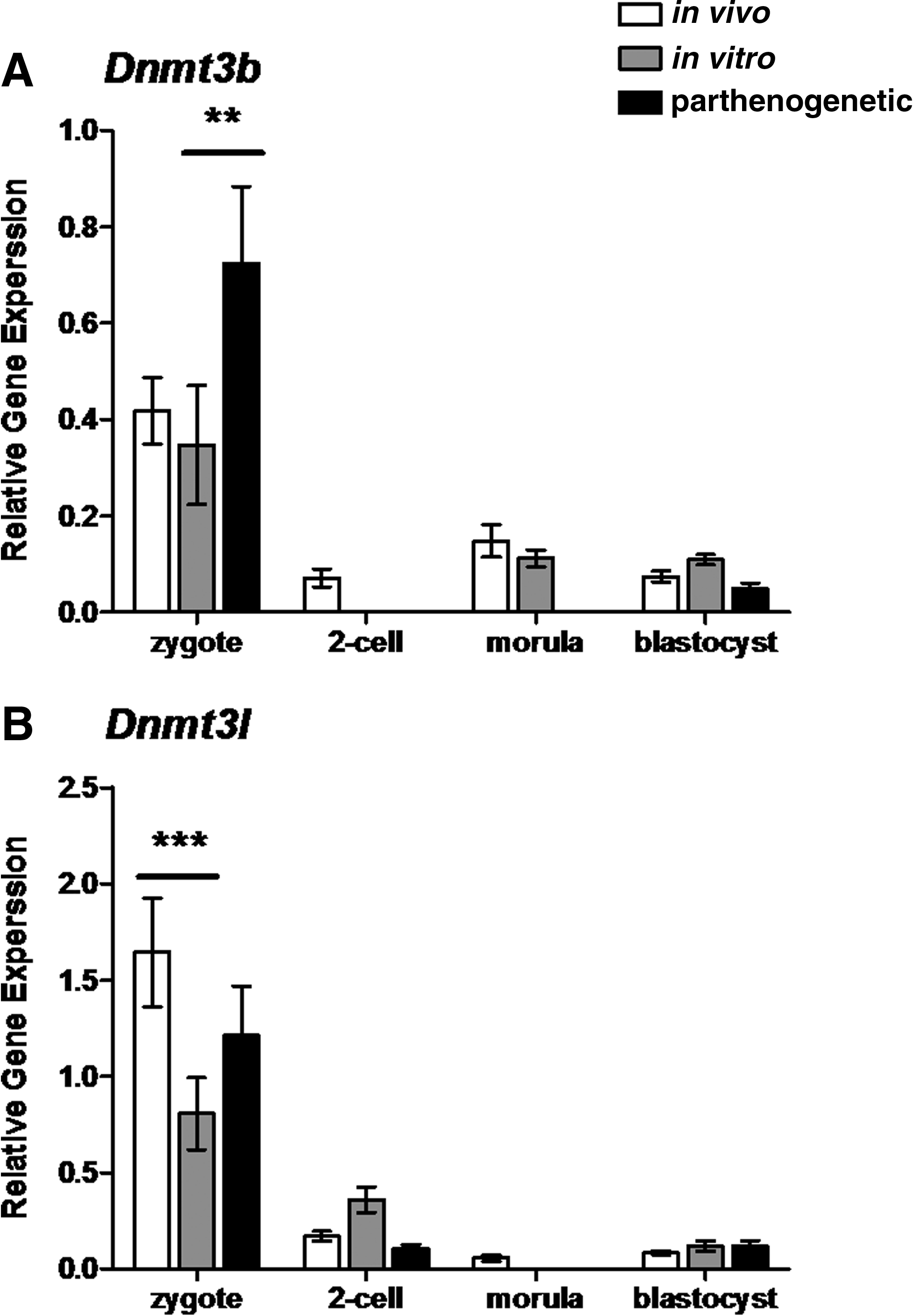
Relative gene expression of the DNA methyltransferase 3B (Dnmt3b) (
In vitro culture and parthenogenesis have opposite effects on the expression of variants of histone 3
Both in vitro manipulation and parthenogenetic activation perturbed expression of the histone variant H3.3b and the histone cell cycle regulation defective homolog A (Hira), the remodeler specific for H3.3b integration (Fig. 4A and B). For in vitro embryos, H3.3b transcript levels were reduced at the zygote stage (two-way ANOVA, p<0.001), whereas parthenogenetic zygotes displayed higher H3.3b levels than in vitro controls (two-way ANOVA, p<0.001) (Fig. 4A). Reduced levels of Hira transcripts were observed within in vitro developing embryos compared to in vivo controls at every assayed developmental stage (two-way ANOVA) (Fig. 4B). Reduced Hira transcripts were also observed for in vitro developing embryos compared to parthenogenetic two-cell, morula, and blastocyst stage embryos (two-way ANOVA) (Fig. 4A and B).

Relative gene expression of the variant of H3 H3.3b (
The in vitro culture of embryos also reduced transcript levels of the centromeric protein A (Cenpa) within the first 24 h of development compared to in vivo embryos (two-way ANOVA, p<0.001) (Fig. 5A). During this time Cenpa transcripts in parthenote zygotes were also higher than in vitro zygotes (two-way ANOVA, p<0.001) (Fig. 5A). Levels of Cenpa were not significantly different between in vivo, in vitro, and parthenogenetic embryos during the subsequent preimplantation stages assayed. In addition, transcript levels of the CENPA-specific remodeler chromatin assembly factor 1 subunit (Caf-1) were not affected by either in vitro culture or parthenogenetic activation at any preimplantation stage assayed (Fig. 5B).

Relative gene expression of the variant of H3, centromeric protein A (Cenpa) (
In vitro culture and parthenogenesis affect gene expression of H2A–H2B dimer constituents
Both in vitro culture and parthenogenesis had an overall effect on polyadenylated H2b transcription (two-way ANOVA, p<0.001 and p<0.01 respectively) (Fig. 6). Polyadenylated H2b transcription was reduced during the first 68 h of development of in vitro cultured embryos relative to in vivo developing embryos (Fig. 6). Zygote levels of H2b in in vitro developing embryos were also lower than in parthenote zygotes, but this difference did not persist into later developmental stages (Fig. 6).
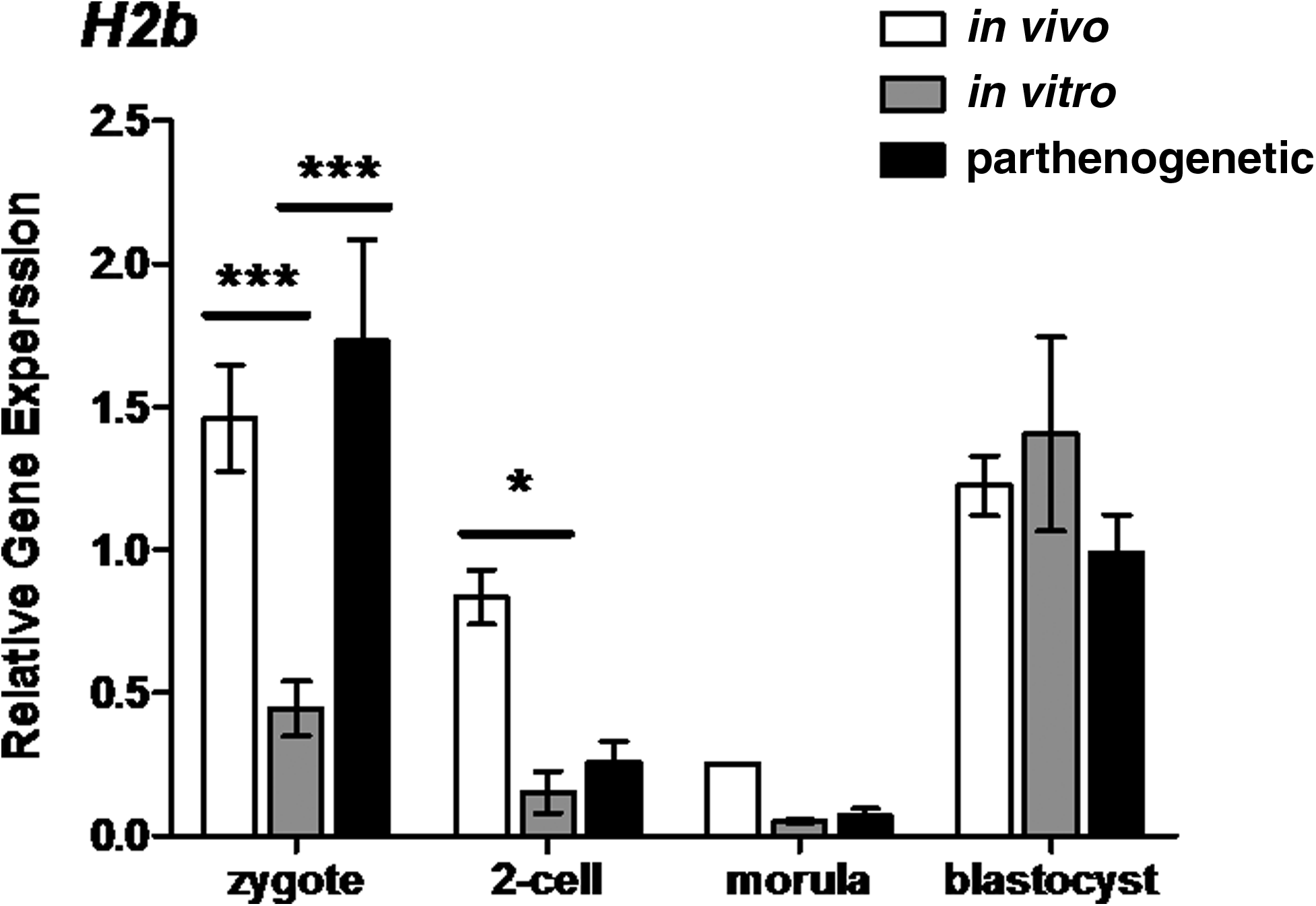
Relative gene expression of the histone H2b in preimplantation mouse embryos. H2b transcript levels were assayed at the zygote, two-cell, morulae, and blastocyst stages (20, 44, 68, and 92 h, respectively). Each column represents mean normalized expression (performed using an endogenous reference gene approach, with the housekeeping gene Rplp0). mRNA was isolated from pools of three embryos (each reaction contains 0.05 embryo equivalent cDNA). Statistical analysis performed between in vivo and in vitro columns and in vitro and parthenogenetic columns using two-way ANOVA with Bonferroni post tests. Graphs are representative of three individual experiments, error bars represent±SEM with significant differences between embryo populations at each stage represented by *(p<0.05) and ***(p<0.001).
The expression of the H2A variant H2afx was reduced in embryos manipulated in vitro. H2afx levels were lower compared to in vivo developing control embryos at every assayed stage except morulae (Fig. 7). H2afx levels in in vitro developing embryos were also lower than parthenogenetic embryo levels at the zygote, two-cell and blastocyst stages (two-way ANOVA, p<0.001, p<0.05, and p<0.05, respectively) (Fig. 7).

Relative gene expression of the histone H2A variant H2afx in preimplantation mouse embryos. H2afx transcript levels were assayed at the zygote, two-cell, morulae, and blastocyst stages (20, 44, 68, and 92 h, respectively). Each column represents mean normalized expression (performed using an endogenous reference gene approach, with the housekeeping gene Rplp0). mRNA was isolated from pools of three embryos (each reaction contains 0.05 embryo equivalent cDNA). Statistical analysis performed between in vivo and in vitro columns and in vitro and parthenogenetic columns using two-way ANOVA with Bonferroni post tests. Graphs are representative of three individual experiments, error bars represent±SEM with significant differences between embryo populations at each stage represented by *(p<0.05), **(p<0.01), and ***(p<0.001).
H2afy was the only H2A variant not affected by in vitro culture or parthenogenesis (Fig. 8). In contrast, both in vitro and parthenogenetic zygotes and blastocysts displayed altered H2afz levels compared to their respective controls (Fig. 9A). At the zygote stage, H2afz expression in in vitro developing embryos was at least threefold higher than in in vivo counterparts (two-way ANOVA, p<0.001). The expression of H2afz in parthenogenetic zygotes was also reduced, to levels at least 10-fold less than those documented for in vitro developing zygotes (two-way ANOVA, p<0.001). At the blastocyst stage, the H2afz expression levels of in vitro embryos were about 80% lower than in vivo embryos (two-way ANOVA, p<0.001). Parthenote blastocysts displayed a sixfold elevation in H2afz levels compared to in vitro blastocysts (two-way ANOVA, p<0.001).
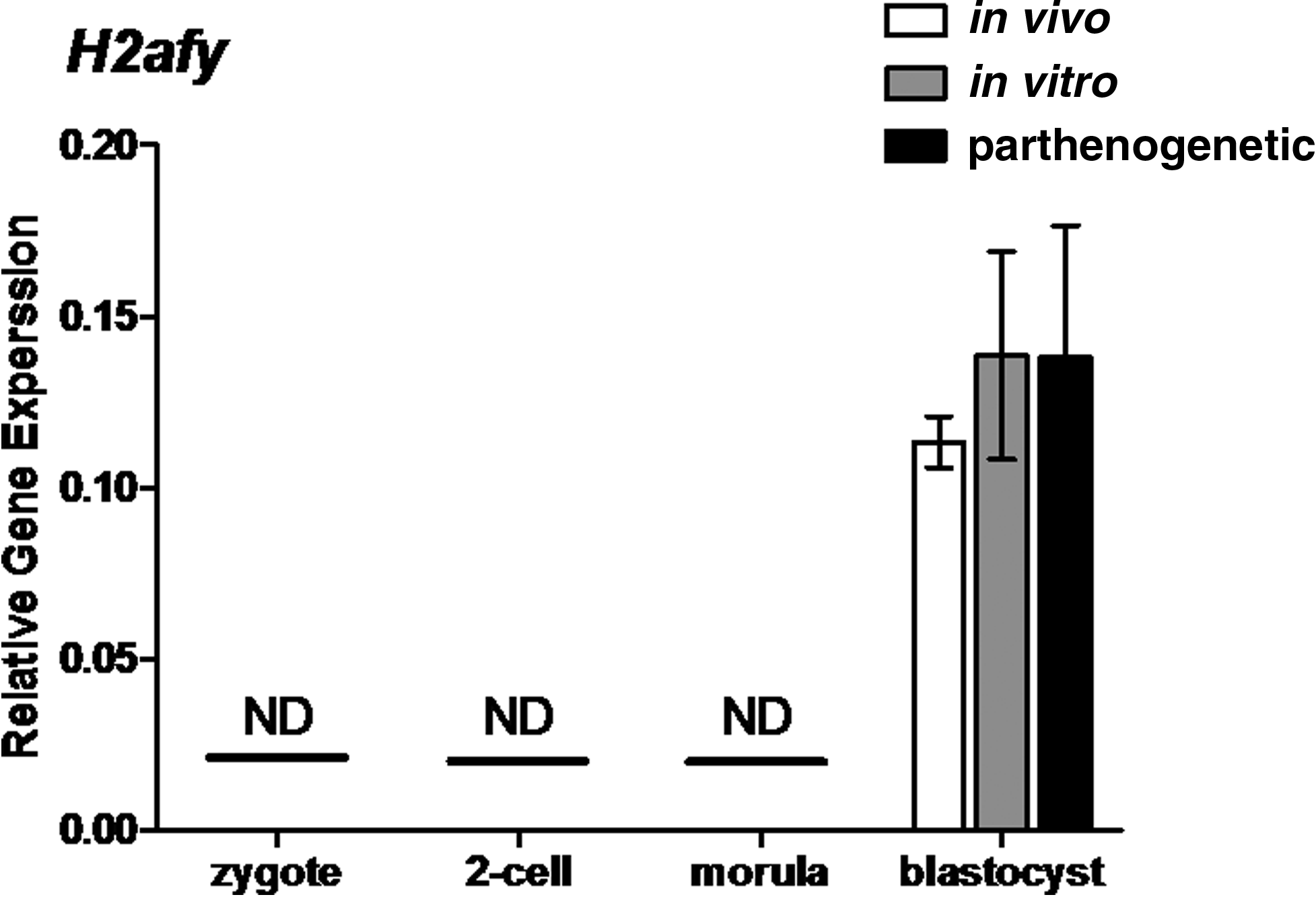
Relative gene expression of the histone H2A variant H2afy in preimplantation mouse embryos. H2afy transcript levels were assayed at the zygote, two-cell, morulae, and blastocyst stages (20, 44, 68, and 92 h, respectively). Each column represents mean normalized expression (performed using an endogenous reference gene approach, with the housekeeping gene Rplp0). mRNA was isolated from pools of three embryos (each reaction contains 0.05 embryo equivalent cDNA). Statistical analysis performed between in vivo and in vitro columns and in vitro and parthenogenetic columns using Student t-tests. Graphs are representative of three individual experiments; error bars represent±SEM. ND represents stages at which transcripts were not detectable by qRT-PCR.
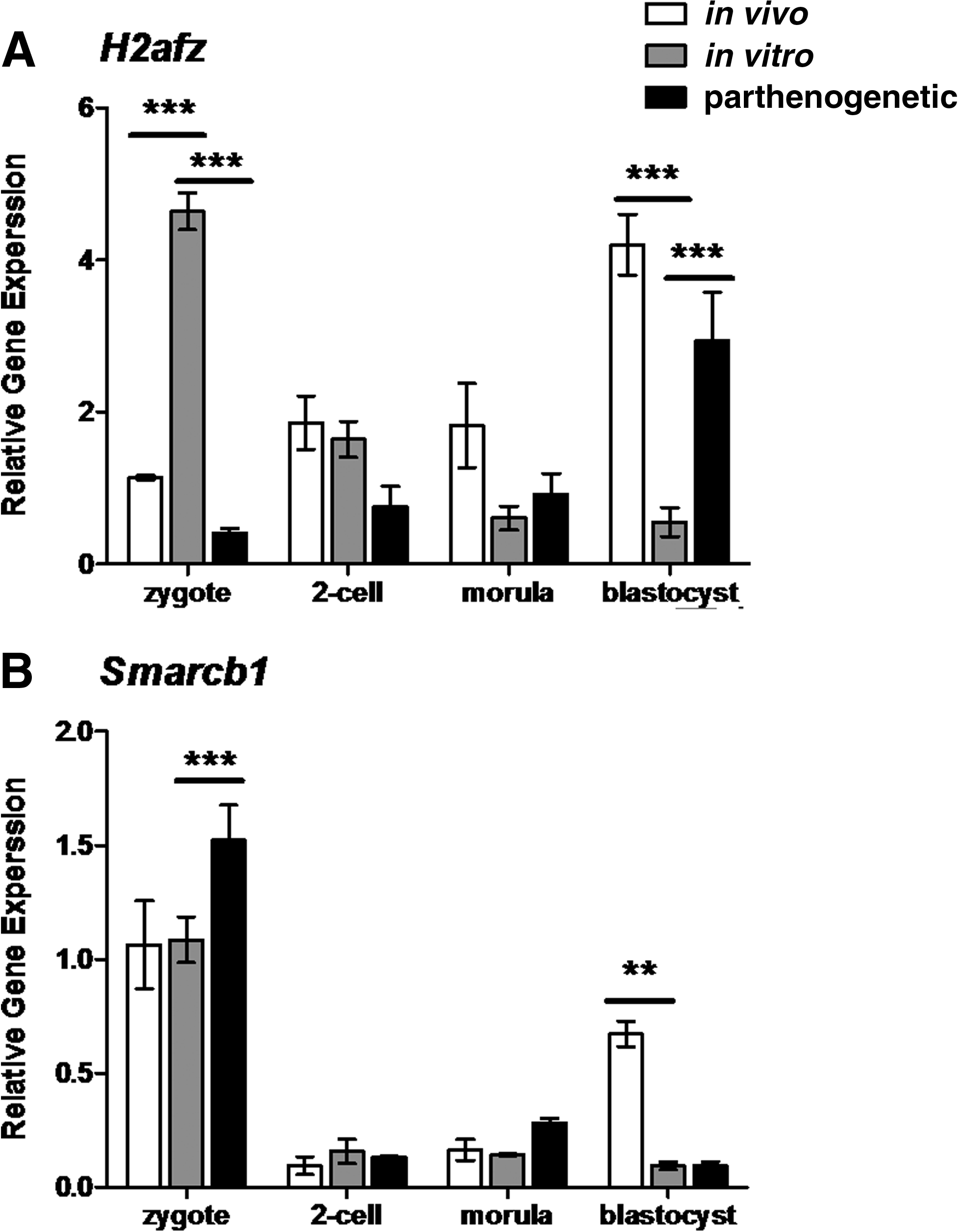
Relative gene expression of the variant of H2A, H2afz (
Expression assays of the B1 subunit of the SWI/SNF related, matrix associated, actin-dependent regulator of chromatin (Smarcb1), the chromatin remodeler specific for H2AFZ somewhat reflected the different expression patterns of embryos of different developmental milieu. Smarcb1 showed that levels were higher in in vivo blastocysts compared to in vitro developing blastocysts (two-way ANOVA, p<0.01) (Fig. 9B). Conversely, Smarcb1 levels were elevated in parthenogenetic zygotes compared to in vivo developing zygotes (two-way ANOVA, p<0.001), but were not different at the blastocyst stage for these embryos (Fig. 9B). Levels of Smarcb1 were not different between in vivo, in vitro, and parthenogenetic two-cell and morulae staged embryos.
To determine if documented mRNA transcript differences were reflected in histone protein changes we also investigated protein levels of H2AFZ in embryos created by in vivo and in vitro means. Western immunoblotting of H2AFZ at the zygote, two-cell, morulae, and blastocyst stages shows more H2AFZ protein in in vitro developing zygotes and two-cell embryos and morulae than in in vivo controls (Fig. 10). Reversible total protein staining with Ponceau S was used in these Western immunoblotting experiments because histone protein extraction precludes the use of typical acid insoluble protein loading controls such as β-ACTIN (data not shown).

Levels of H2AFZ in embryos sampled in vivo or cultured in vitro. Western blot of acid-extracted total protein probed with anti-H2AFZ antibodies. Lanes represent for each type of embryo 30 zygotes (lane Z), 30 2-cells (lane 2C), 30 morulae (lane M), and 20 blastocysts (lane B) (20, 44, 68, and 92 h post-Hcg, respectively). Images are representative of three individual experiments. As reported previously, the isolated H2AFZ protein runs as the bottom band of a doublet at approximately 15 kDa (Kafer et al., 2010).
The expression of genes that code for proteins involved in chromatin environment establishment are typically reduced in in vitro embryos compared to in vivo and parthenogenetic counterparts
The collation of gene expression changes between in vivo and in vitro embryos showed that transcript levels were mostly lower in embryos cultured in vitro compared to in vivo developing controls (Fig. 11). Of the 13 genes assayed, 10 were affected by in vitro culture (Fig. 11). Of these, seven were repressed at the zygote stage (H2b, H2afx, H3.3b, Hira, Cenpa, Hp1, and Dnmt3l), four were repressed at the two-cell stage (H2b, H2afx, Hira, and Hdac1), two were repressed at the morula stage (Hira and Hdac1), and four were repressed at the blastocyst stage (H2afx, H2afz, Smarcb1, and Hira). Only the expression of one gene, H2afz at a single stage, the zygote stage, was elevated relative to in vivo developing control embryos.

Kinetics of histone variant and associated factor expression during development of mouse preimplantation embryos generated from in vivo, in vitro, and parthenogenetic means. Summary of overall expression trends (increased or decreased relative to controls) in a stage by stage, factor by factor manner. In vivo embryos served as control samples for in vitro embryos, and the in vitro embryos served as controls for the parthenogenetic embryos. Embryo stages (zygote, two-cell, morula, and blastocyst) are represented at the bottom of the figure. White boxes represent instances where expression is significantly lower than controls. Gray boxes represent instances where expression is not significantly different to controls and black boxes represent instances in which expression in test group is higher than in control group.
The same genes that were perturbed in comparisons between embryos generated in vivo and in vitro were also perturbed in comparisons between in vitro and parthenogenetic embryos. However, for the latter comparison, a different pattern emerged where, relative to in vitro control embryos, gene transcripts were elevated in parthenotes (Fig. 11). The only exception to this observation was that H2afz expression was reduced in parthenote zygotes relative to in vitro developing zygotes. Of the 13 genes assayed, 10 were elevated in parthenogenetic embryos including 6 at the zygote stage (H2b, H2afx, Smarcb1, H3.3b, Cenpa, and Hp1), 4 at the two-cell stage (H2afx, Hira, Hdac1, and Dnmt3b), 2 at the morula stage (Hira and Hdac1), and 3 at the blastocyst stage (H2afx, H2afz, and Hira).
Discussion
Preimplantation development is by definition an epigenetic system (Santenard and Torres-Padilla, 2009). Given this, this study has focussed specifically on the differential expression of genes involved in maintaining and changing epigenetic chromatin environments in embryos of different developmental milieu. This study was performed to further elucidate if routinely used assisted reproduction technologies may cause changes to the transcription of genes that code for proteins that are directly involved in the establishment of chromatin environments.
Although microarray studies focused on preimplantation staged embryos have in some part improved our understanding of how developmental milieu can impact upon embryonic transcriptomics, these studies require large amounts of input material, which for preimplantation embryos must be achieved by either pooling hundreds of embryos or by amplification (Czechowski et al., 2004; Pfaffl, 2003). Although routinely used, these approaches are not ideal as pooling embryos in groups can obscure individual variations and amplification may introduce bias derived from differential amplification (Bower et al., 2007; Gardner et al., 2004). Through the use of sensitive qRT-PCR assays we have been able to use small pools of three embryos to identify transcriptome changes relating to the chromatin based epigenetic system that have not been previously identified.
Our study confirms that many factors associated with epigenetic chromatin environments are susceptible to gene expression changes as a result of in vitro culture. Our results show that of the genes assayed that were affected by in vitro culture, 90% of these were reduced compared to in vivo controls. Interestingly, however, this phenomenon was not observed in parthenogenetic embryos. Overall, the expression pattern of chromatin based epigenetic constituents in parthenogenetic embryos more closely resembled that of embryos developing in vivo than that of embryos developing in vitro. Other research has suggested that embryos generated through in vitro fertilization (IVF), intracytoplasmic sperm injection (ICSI), and androgenesis suffer from repressed transcriptional activity compared to parthenogenetic counterparts (Bui et al., 2010). Parthenogenetic embryos develop from oocytes that have undergone normal oogenesis and have regenerated a set of naive chromatin without the integration of paternal chromatin (Gao et al., 2007). It has previously been suggested that the in vitro manipulated embryo experiences a repressed chromatin state due to inappropriate remodeling of the paternal chromatin (Burton and Torres-Padilla, 2010). Evidence to support this notion comes from studies of preimplantation development in vivo, which show that maternal chromatin is highly stable and it is the paternal chromatin that is extensively remodelled following fertilization (Burton and Torres-Padilla, 2010). During this time, maternal chromatin remains highly methylated, but paternal chromatin is first decondensed and is then demethylated to induce a transcriptionally repressed state at the two-cell stage (Gao et al., 2007; Ma et al., 2001; Mayer et al., 2000; Young and Beaujean, 2004).
Exactly how in vitro manipulation could impair paternal remodeling is unknown; however, the most likely cause is that repression is due to inappropriate levels of histone acetylation and methylation during or following chromatin reprogramming (Nowak-Imialek et al., 2008; Papp and Muller, 2006; Schotta et al., 2004). Effects on methylation and acetylation levels are likely to be induced by inappropriate activity of the proteins responsible for such covalent modification. In our study, two genes that displayed reduced transcription were genes that code for DNMT and HDAC proteins, the mediators of acetylation and methylation. Both genes families have been shown to be essential for development through embryonic knockdown experiments. HDAC1, a regulator of DNA and histone acetylation (Kanka, 2003) is critical during preimplantation development. A reduction of Hdac1 enhances expression of genes that are usually repressed and results in a lower rate of expanding blastocysts (Ma and Schultz, 2008). Similar to HDAC1 deficient embryos, DNMT depleted embryos also display an impaired differentiative ability. The offspring of female DNMT3L−/− mice perish following hatching and elongation, at approximately 9.5 dpc (Dodge et al., 2004). Although the definitive cause of this embryonic lethality is unknown, aberrant gene expression would occur if methylation of DNA and histone proteins is altered and is therefore a likely cause (Martin et al., 2006; Meehan and Stancheva, 2001).
In addition to documenting aberrant expression of covalent modifiers we have also shown that the in vitro manipulation of mouse preimplantation embryos can cause changes to the levels of transcripts coding for histone proteins. Components of the H2A–H2B dimer and its variants appeared to be largely susceptible to expression changes following in vitro manipulation. H2b expression is high in zygotes when the maternal stores of H2B are used to replace sperm protamines during the extensive chromosome remodelling following fertilization (McLay and Clarke, 1997, 2003). Given this, the early reductions of H2b expression seen in in vitro developing embryos could represent impaired paternal chromatin turnover.
We also document consistent reductions in the expression of the H2A variant H2afx in the in vitro manipulated preimplantation embryo. In embryos developing in vivo, H2afx is one of the highest expressed histone H2A variants (Kafer et al., 2010; Ziegler-Birling et al., 2009). The consistent presence of H2AFX protein during early development is also seen in Xenopus embryos, where H2AFX is the most abundant histone H2A variant (Dimitrov et al., 1994). Because H2AFX plays important roles in the processes that identify and repair damaged DNA, a reduction in H2afx levels could lead to an accumulation of DNA damage (Adiga et al., 2007). Aside from signaling for DNA repair, H2AFX may also have a role in the remodelling of paternal chromatin (Ziegler-Birling et al., 2009). Based on this knowledge, a consequence of reduced H2afx in embryos developing in vitro could be that paternal reprogramming is impaired, which could result in generic transcriptional repression (Paul et al., 2008; Ziegler-Birling et al., 2009).
Our studies show that another H2A variant, H2afy was unaffected by in vitro manipulation or parthenogenetic activation. This is interesting given that H2afy is transcribed from the maternal genome, not paternal genome (Chang et al., 2005) and further supports the hypothesis that paternal chromatin remodelling is primarily effected by in vitro manipulation.
Like H2afx, H2afz is another histone variant that is constantly expressed in in vivo embryos and should peak at the blastocyst stage (Kafer et al., 2010). The results presented here show that the pattern of expression of H2afz and its specific remodeler Smarcb1, differed significantly between in vivo and in vitro developing embryos particularly at the blastocyst stage. H2afz knockdown studies have shown that the aberrant regulation of H2AFZ is detrimental to early mammalian development. H2AFZ knockdown is lethal for mouse preimplantation embryos at the implantation stage, as is the knockdown of the histone remodeler SWI/SNF (of which SMARCB1 is a subunit) (Faast et al., 2001; Klochendler-Yeivin et al., 2000). Perturbation of H2afz levels in the Xenopus embryos also affects differentiation and gastrulation events suggesting that H2AFZ has a specific role in extra-embryonic tissue differentiation (Ridgway et al., 2004). Knockdown studies have also been performed in embryonic stem (ES) cells. The results of these studies indicated that H2AFZ is required for committed cell states not pluripotent states as the pluripotency markers Oct4 and Nanog were elevated in H2AFZ−/− ES cells (Creyghton et al., 2008). Together with our results that show that H2AFZ protein is present in abnormally high levels in in vitro developing embryos we speculate that the cells of embryos developing in vitro may lose their pluripotency earlier than their in vivo counterparts. This is also consistent with the results that show that the derivation of ES cell lineages is more difficult from in vitro embryos than from in vivo or parthenogenetic embryos (Ogawa et al., 2009; Tielens et al., 2006).
Like the H2A variants, our results clearly show that the embryonic levels of H3 variants H3.3b and Cenpa as well as the H3.3 specific remodeler Hira were affected by in vitro manipulation. The role of H3.3B is to establish the early chromatin environment during the zygote and two-cell stage to allow for transcriptional activation (Torres-Padilla et al., 2006). More recently, H3.3B has been shown to specifically establish heterochromatin environments within the paternal genome (Santenard et al., 2010). The reduced levels of H3.3b that we document in the in vitro embryo may be another source for erroneous establishment of paternal chromatic environments.
This study has provided new information which contributes to ongoing research aimed at identifying the possible epigenetic impacts of in vitro-based reproductive technologies. DNA and histone covalent modification as well as the replacement and reorganization of chromatin environments and histone octamer are only some aspects of the broader epigenetic system that regulates development. Although we show that some aspects of chromatin-based epigenetics are not influenced by different developmental milieu, many aspects are, including critical modifiers of acetylation and methylation and histone variants such as H2afz, H2afx, and H3.3b. We also generate further support for the hypothesis that mammalian embryos cultured in vitro are more likely experience transcriptional repression, at least in genes that are linked to the epigenetic remodeling of chromatin environments. Further, we show that this repression does not occur in parthenogenetic embryos; reinforcing the hypothesis that paternal chromatin reorganization is a potential source for erroneous reprogramming in in vitro cultured embryos.
Footnotes
Acknowledgments
Georgia Rose Kafer is a recipient of an Australian Post-Graduate Award and a CSIRO Livestock Industries Scholarship.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.