
Abstract
Abstract
Blood cells transfusion and hematopoietic stem cells (HSCs) transplantation are important methods for cell therapy. They are widely used in the treatment of incurable hematological disorder, infectious diseases, genetic diseases, and immunologic deficiency. However, their availability is limited by quantity, capacity of proliferation and the risk of blood transfusion complications. Recently, human embryonic stem cells (hESCs) have been shown to be an alternative resource for the generation of hematopoietic cells. In the current study, we describe a novel method for the efficient production of hematopoietic cells from hESCs. The stable human fetal liver stromal cell lines (hFLSCs) expressing erythropoietin (EPO) were established using the lentiviral system. We observed that the supernatant from the EPO transfected hFLSCs could induce the hESCs differentiation into hematopoietic cells, especially erythroid cells. They not only expressed fetal and embryonic globins but also expressed the adult-globin chain on further maturation. In addition, these hESCs-derived erythroid cells possess oxygen-transporting capacity, which indicated hESCs could generate terminally mature progenies. This should be useful for ultimately developing an animal-free culture system to generate large numbers of erythroid cells from hESCs and provide an experimental model to study early human erythropoiesis.
Introduction
Mok et al. (2008) have shown that the supernatants from human mesenchymal stromals cells (MSCs) transfected with erythropoietin (EPO) could induce the differentiation of HSCs into erythroid colonies. It was also shown that direct secreted cytokines from stromal cells exert their function better than medium supplement (Abboud et al., 1998). Thus, we hypothesized that such an inducing system could be used to promote hematopoietic differentiation of hESCs. Hematopoiesis in human is regulated both temporally and spatially in a dynamic manner. The hematopoietic system develops from the aorta-gonado-mesonephros (AGM) region, then migrates to the fetal liver, and finally shifts to the bone marrow. The fetal liver is a unique hematopoietic microenvironments that facilitate both HSCs and mature blood cells generated (Takeuchi et al., 2002). In the present study, we established human fetal liver stromal cells (hFLSCs) overexpressing EPO protein. The conditioned medium (CM) from this stromal cells promoted hESCs differentiation into various types of hematopoietic progenitor cells, including meyeloid, erythroid, and multipotential progenitors, especially erythroid cells.
Materials and Methods
Isolation of human fetal liver stromal stem cells (hFLSCs)
This work was approved by the Ethics Committee of Chinese PLA General Hospital. Fetal tissue collection complied with international guidelines regarding the use of fetal tissue for research (Lindskog et al., 2006). The human fetal liver samples were collected from normal aborted fetal tissue of 14 weeks (Liu et al., 2010). Written informed consent was obtained from the voluntary pregnant women who donated the fetal tissue for this research purposes. hFLSC were isolated as previously described (Liu et al., 2009; Sato et al., 2003; Zhao et al., 2008). Briefly, samples of human fetal liver were minced into 1 mm3 pieces, and then cultured in medium consisting of 45% Dulbecco's modified Eagle's medium (DMEM), 45% DF12, and 10% fetal bovine serum (FBS; Hyclone, Logan, UT). The tissue pieces were incubated undisturbed for 1 week at 37°C in a humidified atmosphere with 5% CO2 in air, allowing the cells to migrate and adhere to the flask surface. The adherent cells were harvested by digesting cells in cocultures with 0.25% trypsin/0.53 mM EDTA solution and reseeded into 25 cm2 cell culture flasks. One week after confluence, the hFLSC feeders were established and frozen in liquid nitrogen, within five generations.
The establishment of EPO-expressing hFLSC
The human EPO complete coding sequence (598 bp, GenBank accession number: NM_000799) was cloned from the human fetal liver tissue using RT-PCR. Then the amplified human EPO fragments was ligated into a pBPLV lentiviral vector plasmids containing eGFP (kindly provided by Dr. Luigi Naldiai, Vita Salute San Raffaele University, Italy). The lentiviruses were assembled in HEK293T cells lines (Invitrogen, Carlsbad, CA). The viruses were used to infect the hFLSCs supplemented with 4 μg/mL polybrene (Sigma, Oakville, ON, Canada). After 48 h of transduction, the eGFP-positive hFLSCs that constitutively expressed EPO were sorted by fluorescence-activated cell sorting (FACS). The capacity of cell proliferation was determined as previously described (Itano et al., 2002).
Western Blot to assay the expression of EPO protein in hFLSCs
The hFLSCs with or without the stable expression of EPO gene and those transfected with empty vector were analyzed for the production of recombinant EPO protein by Western blotting. Proteins were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to a polyvinylidene difluoride (PDVF) membranes (Milipore, Bedford, MA). After blocking in 5% skimmed milk the membranes were incubated with the anti human EPO antibody (R&D, Minneapolis, MN), followed by peroxidase-conjugated secondary antibodies. The protein bands were visualized by enhanced chemiluminescence (Santa-Cruz, Santa Cruz, CA).
Determination of EPO expression by enzyme-linked immunosorbent assay (ELISA)
ELISA was performed using human Quantikine kits for secreted protein EPO (R&D), according to the manufacturer's recommended protocol. Proteins extracted from culture supernatants were standardized for total protein content using the BCA Protein Assay kit (Pierce, Rockford, IL) before analysis. The results represent the outcome of three independent experiments.
Conditioned medium
The hFLSCs with or without EPO transfection were cultured in a medium comprising 45% DMEM, 45% DMEM/F12, and supplemented with 10% FBS at 37°C in a humidified atmosphere flushed with 5% CO2 in air. The hFLSCs within five generations were irradiated (20 Gy) and dissociated with 0.25% trypsin/0.53 mM EDTA, and then replated onto six-well cell culture plates at the density of 2×105 per well. On the following day, the cultures were washed in phosphate-buffered saline (PBS) and with medium consisting of IMDM and 10% FBS. Conditioned medium was collected daily, filter-sterilized and stored at −20°C.
Culture of human ES cells
Human embryonic stem cell line H9 was purchased from WiCell Research Institute (Madison, WI). The hESCs were routinely culture on irradiated mouse embryonic fibroblast (MEF) feeder cells in knock-out DMEM (Invitrogen) containing 20% serum replacer (SR; Knockout SR, Invitrogen), 1% nonessential amino acids (NEAA), 1 mM L-glutamine (all from Invitrogen), 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO) and 4 ng/mL human basic fibroblast growth factor (bFGF) (R&D). The hESCs were subcultured approximately every 6 days using collagenase IV (1 mg/mL, Gibco Laboratories, Grand Island, NY), and the passage number of H9 cells used in this paper was between 40 and 65.
MEF cells were grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin.
Formation of embryoid bodies (EBs)
For EBs formation, undifferentiated hESCs were harvested at confluence with collagenase IV and scraped off in strips. They then were transferred to low-attachment plates to allow for EBs formation by overnight incubation in differentiation medium consisting of knock-out DMEM supplemented with 20% FBS, 1% NEAA, 1 mM L-glutamine, and 0.1 mM β-mercaptoethanol in the presence of 25 ng/mL rhBMP4 for 24 h (Zhang et al., 2008). On the next day, the EBs were divided into three groups: (1) hFLSCs-CM, (2) hFLSCs-CM+2 U/mL EPO, (3) EPO/hFLSCs-CM.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen) as described by the manufacturer's protocol. RNA was then reverse-transcribed into cDNA by MMLV reverse transcriptase (Promega, Madison, WI). PCR amplification of different genes was performed with rTaq polymerase (Takara Inc., Otsu Shiga, Japan). Cycle conditions were as follows: 94°C for 5 min followed by 30 cycles (94°C denaturation for 30 sec, 53–61°C annealing for 30 sec, 72°C extension for 30 sec) with a final incubation at 72°C for 10 min. The primers used in the amplification are shown in Table 1.
F, forward; R, reverse; bp, base pair.
Real-time RT-PCR
The gene expression of EBs was analyzed with real-time RT-PCR using the SYBR Green PCR Master Mix following the manufacturer's instructions. Total RNA in different groups was extracted on days 2, 4, 6, and 8. All PCR reactions were performed as follows: 94°C for 5 min and cycled at 94°C for 30 sec, 52–62°C for 30 sec, and 72°C for 30 sec (35 cycles) followed by final extension at 72°C for 10 min, 4°C for 5 min. The primers used in the detection were shown in Table 2.
F, forward; R, reverse; bp, base pair.
Flow cytometric analysis
The trypsinized individual cells were incubated with the FITC-conjugated and PE-conjugated monoclonal antibodies: antihuman CD29, antihuman CD105, antihuman CD44, antihuman CD90, antihuman CD34, and antihuman CD45 (BD Biosciences, San Jose, CA) at 4°C for 30 min. Then the cells were washed three times with PBS and analyzed by flow cytometry analysis using the FACSCalibur (Becton-Dickinson, Mountain View, CA).
Hematopoietic colony culture
Hematopoietic colony assays were performed in 35-mm low-adhesion plastic dishes using MethoCult GF-H4434 semisolid medium (Stem Cell Technologies, Vancouver, Canada) consisting of 1% methylcellulose, 30% FBS, 1% bovine serum albumin (BSA), 50 ng/mL stem cell factor, 20 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF), 20 ng/mL granulocyte colony-stimulating factor (G-CSF), 20 ng/mL interleukin-3 (IL-3), 20 ng/mL interleukin-6 (IL-6), and 3 U/mL EPO. After culturing for 12–14 days at 37°C and 5% CO2 in a humidified atmosphere, colonies were scored according to their cellular morphology characteristics.
Wright-Giemsa staining and benzidine staining
To analyze hESC-derived erythroid cell morphology, colonies were individually isolated from methylcellulose by aspiration and washed in PBS. Cells were dropped onto slides and fixed for 20 min in 4% paraformaldehyde. The smears were stained with Wright-Giemsa reagents (Fisher Scientific, Fairlane, NJ) and 3′3-diaminobenzidine reagent (Sigma-Aldrich) according to the manufacturer's instructions.
Functional assays of hESC-derived erythroid cells
To detect G6PD activity, WST-8 method was used according to the manufacturer's instructions (Kumamoto, Japan) (Arai et al., 2006). The oxygen binding ability of hESC-derived erythroid cells and human CB was measured with a Hemox analyzer, as previously described (Honig et al., 1990; Shirasawa et al., 2003).
Statistical analysis
Results were expressed as means±SEM. Statistical significance was determined using an unpaired Student t-test. Results were considered significant at p<0.05.
Results
Overexpression of EPO in hFLSCs
We isolated and cultured hFLSCs from 14 weeks human fetal liver. After the primary and two subsequent passages, hFLSCs were adherent to be fusiform shape and highly uniform in morphology (Fig. 1A). We constructed a pBPLV-EPO vector with enhanced green fluorescent protein (eGFP) to increase the expression level of EPO protein in hFLSCs. We found that the transfected hFLSCs maintained a growth rate similar to the untransduced hFLSCs (Fig. 1B). The Egfp-positive expression cells were sorted by FACS. After sorting the transduced hFLSCs showed high expression of eGFP (Fig. 1C). The phenotype of hFLSCs with or without transgene was essentially free of adipocytes, hematopoietic cells, and endothelial cells (CD34− and CD45−), whereas they exhibited markers unique to stromal cells such as human mesenchymal stromal cells (hMSCs), including CD44, CD29, CD105, and CD90 (Fig. 1D).
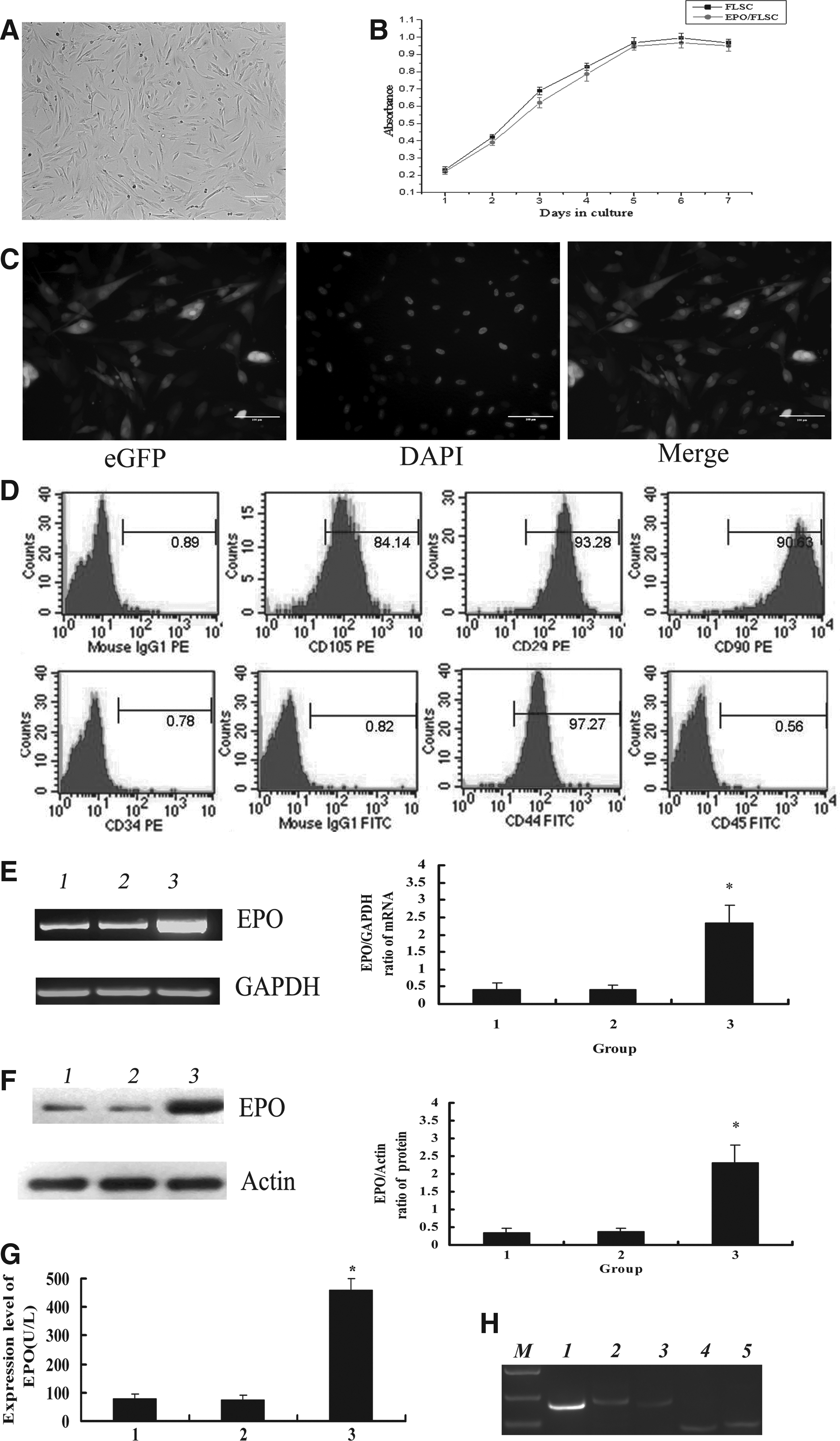
Morphology and identification of EPO overexpressing hFLSCs. (
We collected the sorted transduced hFLSCs and examined the relative expression level of EPO genes by RT-PCR and Western blotting. The data showed that EPO mRNA in EPO/hFLSCs was much higher than in hFLSCs without transfection or transfected with empty vector (p<0.05) (Fig. 1E). The level of EPO protein in transduced cells was higher than that of hFLSCs without transfection or transfected with empty vector (p<0.05) (Fig. 1F). The expression level of EPO in the hFLSCs was examined by ELISA. The results showed that EPO protein levels in EPO/hFLSCs ranged from 263 to 459U/L (data not shown), and were higher than in the untransfected hFLSCs or the hFLSCs transfected with the empty vector (p<0.05) (Fig. 1G). These data proved that the EPO protein was overexpressed in the transduced hFLSCs. Furthermore, the expression of cytokines from EPO/hFLSCs were analyzed. The EPO/hFLSCs expressed detectable levels of EPO receptor (EPOR), SCF, SDF-1, and IL-6 (Fig.1H).
EPO/hFLSCs-CM Promotes Hematopoietic Development of Human Embryoid Bodies
To achieve EBs formation, hESCs were transferred to low-attachment plates in the presence of BMP4. The typical round-shaped EBs formed after 48 h (Fig. 2A). The cells from different groups were tested for expression of CD34 (the marker for hematopoietic stem cells) at different induction time points. For EBs treated with hFLSCs-CM, CD34+ cells were first detected on day 3, and their number peaked to 10.57% on day 12. CD45+ cells could be detected at day 6 and peaked to 10.06% at day 14. For the EBs treated with hFLSCs-CM+EPO, CD34+ cells could be first detected at day 3, with their number reaching the maximum of 13.1% at day 12. CD45+ cells were first found at day 5 and peaked to 16.14% at day 14. For the EBs treated with EPO/hFLSCs-CM, CD34+ cells emerged before day 2, increased on day 4 of culture, and rised to 22.74% at day 12. C45+ cells were first found with small numbers at day 3 and the number of them appears to be greatly increased at days 12–14 (Fig. 2B). These data demonstrated that EPO/hFLSCs-CM-treated EBs could yield much more hematopoietic cells than other groups.
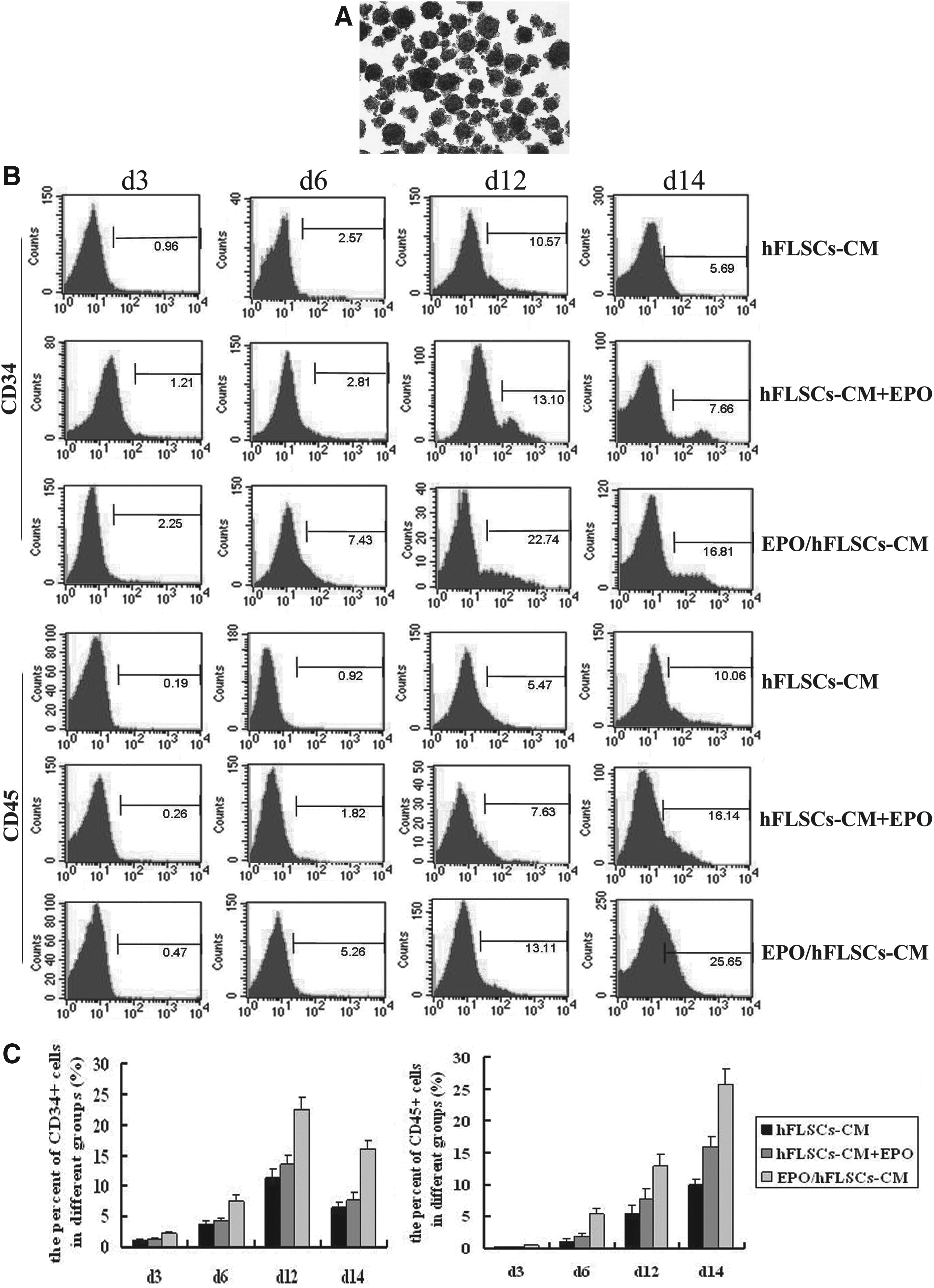
Flow cytometry analysis of hematopoietic differentiation of EBs. (
Evaluation of the hematopoietic potential of the EBs is defined by multilineage hematopoietic capacity using in vitro colony forming assay. Colonies were scored by gross morphology to be hematopoietic were examined for commitment and maturation. When EBs treated with hFLSCs-CM, fewer colonies could be found during the 8-day culture. In the case of hFLSCs-CM+EPO-treated EBs, hematopoietic colonies were first found in day 4 EBs. In addition, there was no significant difference in the number of colonies produced from the two groups. For EPO/hFLSCs-CM-treated EBs, hematopoietic colonies were first found in day 2 EBs, and the number of them was much numerous than the other two groups (Fig. 3A). The number of CD34+ cells and number of colony-forming cells (CFCs) are quantified under the different culture conditions between day 2 and 8 of differentiation. For EPO/hFLSCs-CM treated EBs, the number of CD34+ cells and number of CFCs was much more than other groups (Table 3). This observation demonstrated that EPO/hFLSCs-CM treatment could promote hematopoietic differentiation of the EBs.
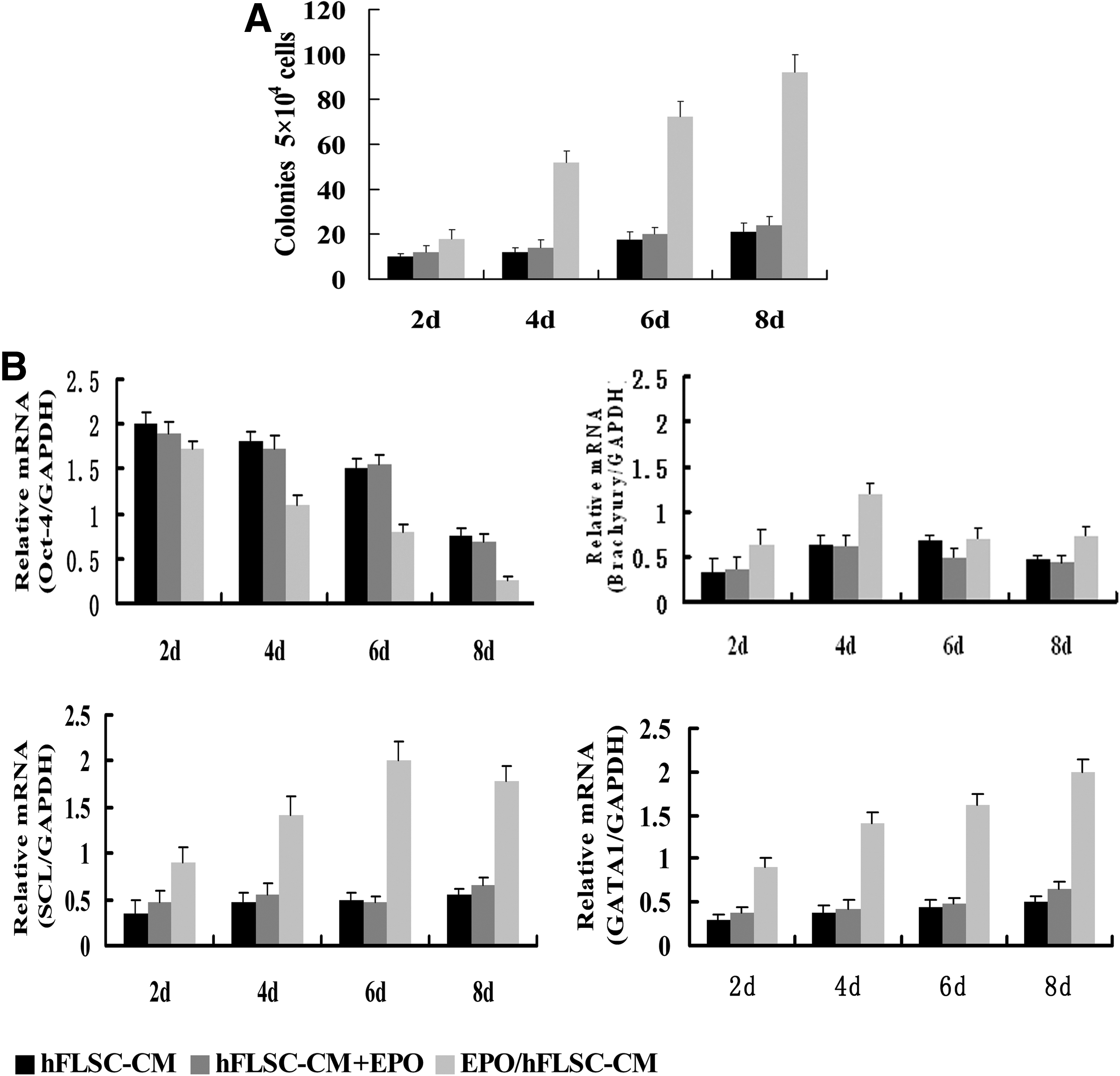
The hematopoietic progenitor development and gene expression of hEBs in different groups. (
We analyzed the gene expression of EBs after different inducing times and found that EPO/hFLSCs-CM could promote the hematopoietic gene expression of EBs. For the hFLSCs-CM-treated EBs and hFLSCs-CM+EPO-treated EBs, Oct-4 (the core transcription factor for regulating self-renewal and survival of hESCs) was expressed during 8 days of differentiation, and then the levels of expression decreased. Brachyury (a marker of primitive streak cells and nascent mesoderm) was detected 2 days later, and SCL (the hematopoietic specific gene) was first detected at 4 days of culture; these markers were present throughout the culture. GATA1 (the initial erythroid-associated transcripts) were maintained at a low level in cultured cells. For the EPO/hFLSCs–CM-treated EBs, Oct-4 expression decreased greatly and declined to undectectable levels at 6 days culture, and Brachyury peaked on days 3 and then declined to undetectable levels by day 6. SCL was first detectable at day 2 and upregulated from day 4. The expression of these hematopoietic-associated genes persisted at relatively high levels throughout the culture (Fig. 3B).
Generation of clonal erythroid progenitors from hEBs
To confirm that the cells induced from EBs could develop into erythroid progenitors, we seeded 1×105 cells in methylcellulose to characterize functionally the frequency of erythroid progenitors after 14 days of coculture. For three groups, the hematopoietic-like colonies arised after 6 days of culture, the erythroid-CFCs was first found at day 8 and then rapidly increased in number, peaked at day 14. The frequency of erythroid precursors produced by EPO/hFLSCs-CM treatment was higher than the in other groups (Fig. 4A). The erythroid cell colonies including erythroid (E) colonies and E bursts were observed (Fig. 4B–E) and E-CFUs appeared later than E burst-forming cells (E-BFUs). Figure 4F showed erythroid cells from one hESC-derived E-BFUs.

Clonogenic growth of hEBs. (
To continue the characterization of the clonal erythroid progenitors obtained from EBs, Wright-Giemsa staining was performed. The morphology of erythroid colonies (Fig. 4G) indicated they were mainly fetal-like definitive erythroid blasts. The erythroid cells from E-BFUs were stained with benzidine (Fig. 4H).
We also analyzed globin gene expression in individual E-BFUs by RT-PCR (Supplementary Fig. 1; see online supplementary data at www.liebertonline.com/cell). They all expressed α-, γ-, β-, ɛ-globin, but ζ-globin was expressed only in four of five of E-BFUs.
Functional assays of hESC-derived erythroid cells
In the previous experiment, we found that EPO/hFLSCs-CM was the best method to induce the hematopoietic development of EBs in three groups, so EPO/hFLSCs-CM-treated EBs were used for the following experiment described below. Human CB-derived erythroid cells were used as a control.
To assess the function of the hESC-derived erythroid cells, we measured oxygen dissociation. The hESC-derived erythroid cells displayed an oxygen dissociation pattern similar to that of human CB (Fig. 5A). These results showed that the function of hESC-derived erythroid cells are similar to fetal RBCs. We also examined glucose-6-phoshpate dehydrogenase (G6PD) activity to confirm that their capability to act against oxidative damage is comparable to that of human CB derived erythroid cells (Fig. 5B).
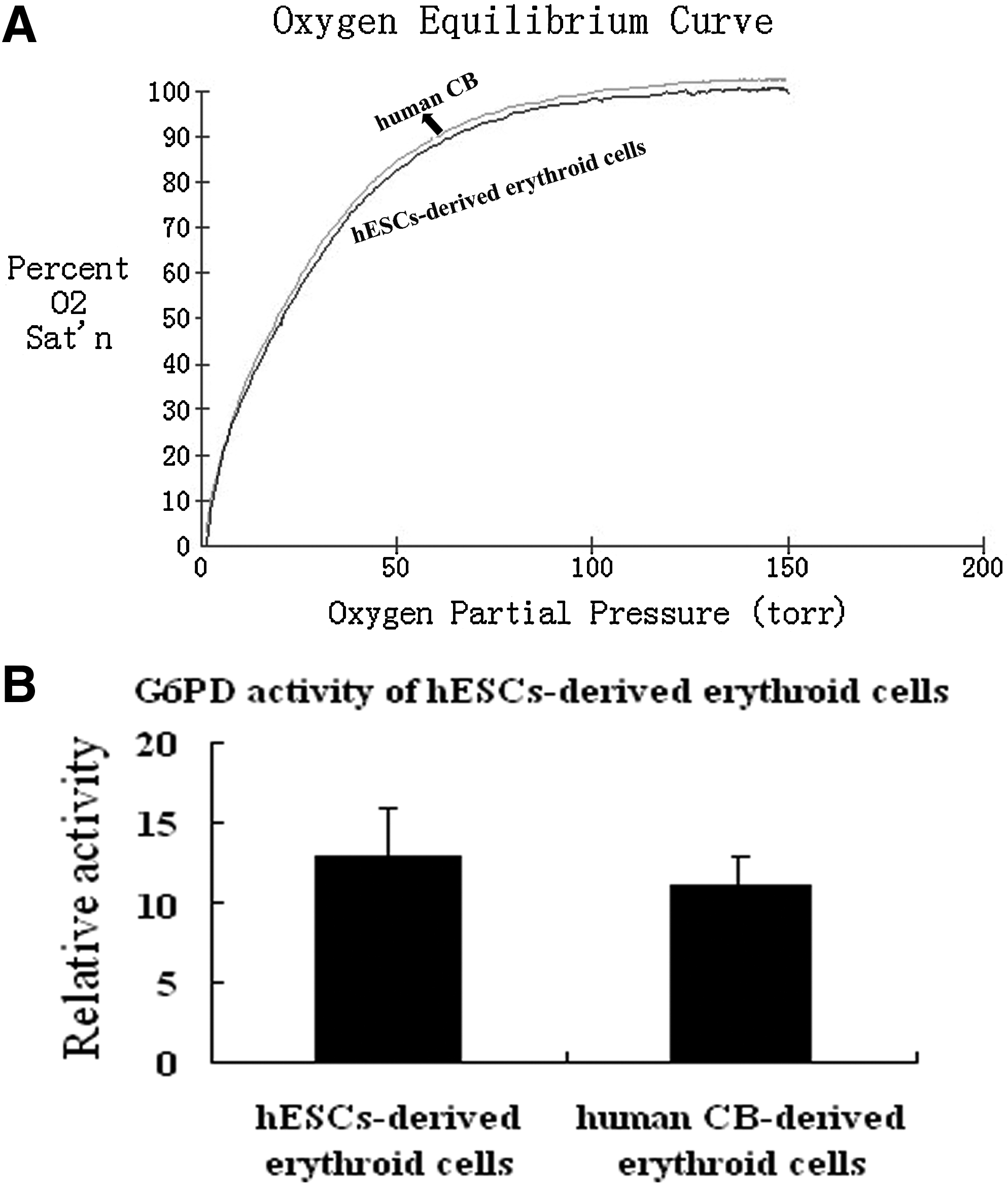
Clonal analysis of hESC derived erythroid cells. (
Discussion
Studies of hematopoietic differentiation of human embryonic stem cells use three main approaches: coculture with variety of hematopoietic supportive stromal cell layers, formation of EBs with cytokines, or a combination of both (Kaufman et al., 2001; Kennedy et al., 2007; Tian et al., 2008; Vodyanik et al. 2005). Thus far, most of the stromal cell lines used to induce hematopoietic differentiation of hESCs have been of murine bone marrow origin. Several studies report that hESCs could differentiate into hematopoietic cells through EBs formation and treatment, where the risk of mouse-related disease should be decreased. However, the low differentiation efficiency of hESCs would greatly limit the future clinical applications. Consequently, defining an inducing system that will safely and consistently drive differentiation of hESCs is urgent. Recent evidence shows that the fetal liver derived stromal cell could induce the hematopoietic differentiation of hESCs (Ma et al., 2007; Wang et al., 2005). The fetal liver is the principal hematopoietic organ where both HSCs and mature blood cells are actively generated and EPO is the principal factor in the regulation of erythropoiesis. Therefore, we attempted to establish the transgenic human fetal liver stromal cells (EPO/hFLSCs) that stably express EPO gene and to use a conditioned medium to induce erythropoietic differentiation of hESCs.
In the current study, we have established a new method to induce significant hematopoietic cells (especially erythroid cells) from hESCs by using conditioned medium from EPO/hFLSCs. In the presence of this conditioned medium a greater number of erythroid colonies were generated from hEBs than in other groups. The hESC-drived erythroid were also able to function as an oxygen carriers, and displayed oxygen dissociation curve similar to human CB. Furthermore, the hESC-derived erythroid cells from this conditioned medium also showed high G6PD activity comparable to that of human CB-derived erythroid cells. These results demonstrated that the hESC-derived erythroid cells had oxygen-carrying properties, but significant effort in bioprocess engineering are still needed to make clinical applications available.
In conclusion, we established the transgenic human fetal liver stromal cells (EPO/hFLSCs) that stably express EPO gene and used the conditioned medium of EPO/hFLSCs to induce erythropoietic differentiation of hESCs. In addition, this method avoided the mouse-related disease and cut down the cost of experiment. The system will provide a reliable alternative for the research in the future therapeutic applications of human embryonic stem cells.
Footnotes
Acknowledgments
We thank Dr. Luigi Naldiai (Vita Salute San Raffaele University, Italy) for the gift of pBPLV lentiviral vector plasmids. Grant sponsor: National High Technology Research and Development Program of China; Grant number: 2006AA02A107. Grant sponsor: the Major State Basic Research Program of China; Grant number: 2005CB522702. Grant sponsor: The Project of Beijing Municipal Science & Technology Commission; Grant number: Z0005190043331.
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.