
Abstract
Abstract
Treatment with cytoplasmic extracts from Xenopus laevis eggs represents a potential tool for universal cellular reprogramming. However, the biochemical activity and quality of the extract vary from batch to batch. This study aimed to evaluate three different extract batches prepared by the same method based on the colony formation of cells after extract treatment, and subsequent in vitro cloning efficiency using treated cells as chromatin donors. Porcine fetal fibroblasts were treated with each batch of extract, and cultured in embryonic stem cell (ES) medium for 12 days. The number of forming colonies in treated cells was counted on Day 7 after extract treatment and significant variability was detected between different batches of extract. Similarly, when using cells from colonies at Days 7 to 8 after treatment for handmade cloning, increased blastocyst formation rates were observed after the cells were treated with a batch showing higher colony formation. In conclusion, assessment of cell colony formation may be used as selection marker for Xenopus egg extract used for pretreatment of donor cells prior to cloning.
Introduction
However, each extract batch is varying in its biochemical activity (Allen et al., 2007; Miyamoto et al., 2008), and there is no clearly defined method to characterize a well-functioning extract. Some standard physical measurments, such as protein concentration and pH, were used in some studies (Bru et al., 2008; Wolffe and Schild, 1991), but most publications represent only random selection of extracts prepared from high-quality eggs.
Colony formation is one of the basic morphological markers used for characterization of stem cells (Shamblott et al., 1998) and induced pluripotent stem (iPS) cells (Ezashi et al., 2009). Colony formation has also been observed both in adherent fibroblasts (Miyamoto et al., 2007) and suspended blood leukocytes (Hansis et al., 2004) after Xenopus egg extract treatment, which may indicate some characteristics of cells have been changed by the extract. After forming an aggregated colony, the cell–cell communication might be increased, which may result in a heterogeneous environment with and between surrounding cells of the colony (Nagaoka et al., 2006). Hence, the changes of characteristics, like growth pattern, in cells treated by Xenopus egg extract might indicate the biological activity of the extract.
Beside morphology, pluripotent cells also display typical gene expression pattern, and pluripotency marker's expression; for example, OCT4, NANOG, and SOX2 (Ezashi et al., 2009; Taranger et al., 2005) can be used to evaluate reprogramming methods and to define pluripotent cell lineages. Expression of OCT4 and SOX2 are the essential markers for maintaining pluripotency of ES cells (Niwa, 2007) and for establishing iPS cells (Ezashi et al., 2009). On the other hand, E-Cadherin (CDH) is reported to be essential for intercellular adhesion, colony formation, and differentiation of ES cells (Chou et al., 2008; Larue et al., 1996), and it is also critical for the generation of iPS cells (Chen et al., 2010).
It has been shown that pretreatment of donor cells with extract from Xenopus oocytes and eggs can increase cloning efficiency in both ovine (Rathbone et al., 2010) and porcine (Liu et al., 2011). Therefore, the aim of this study was to investigate the effect of pretreatment of porcine donor cells with different batches of Xenopus egg extract on cloning efficiency, shown as blastocyst formation, and to evaluate if colony formation induced by extract during cell culture may be used as an assessment marker for increasing the cloning efficiency when extract-treated cells were used as cell donors in nuclear transfer.
Materials and Methods
All chemicals were purchased from Sigma Chemical Co. (St. Louis, MO), unless otherwise stated.
Preparation of somatic cells
Porcine fetal fibroblast monolayer cultures were established from a 40-day-old fetus as described previously (Kragh et al., 2004). Cells grown in Dulbecco's Modified Eagle Medium (DMEM; Gibco BRL, Life Technologies, Grand Island, NY) supplemented with 10% fetal calf serum (FCS, Gibco BRL, Paisley, UK) at passage 3–12 were used for extract treatment and hand-made cloning (HMC).
Preparation of Xenopus egg extract
Xenopus egg extract were prepared as described previously (Liu et al., 2011). Briefly, freshly laid eggs were collected from superovulated Xenopus frogs. The eggs' jelly coat was removed by dejelly buffer (1 mM DTT, 20 mM Tris-HCl, and 100 mM NaCl) for 4 to 5 min, and washed in 0.25× Mare's Modified Ringers (MMR; 100 mM NaCl, 2 mM KCl, 1 mM MgSO4, 2 mM CaCl2, 0.1 mM EDTA, and 5 mM HEPES, pH 7.8) to remove the remaining jelly cover. Eggs were then rinsed with extraction buffer. After removal of excessive buffer, eggs were centrifuged at 15,000×g for 30 min at 4°C. The middle cytoplasmic layer of the tubes were collected and recentrifuged at 15,000×g for 20 min at 4°C. The cleared cytoplasmic extract was supplemented with 5% glycerol, frozen in liquid nitrogen, and stored at −80°C. For this study, three different batches of extracts were prepared from three frogs by the same method. Protein concentration of each batch extract was measured by Nanodrop.
Xenopus egg extract treatment and in vitro cell culture
Fibroblasts grown on poly-L-lysine-coated coverslips (30,000–40,000 cells in each well of four-well dishes) (Nunc, Naperville, IL) were first permeabilized in Hank's balanced salt solution containing 7 μg/mL digitonin on ice for 2 min (Liu et al., 2011). Cells were then incubated in extract containing an ATP-regenerating system (2.5 mM ATP, 125 μM GTP, 62.5 μg/mL creatine kinase, and 25 mM phosphocreatine, as well as 1 mM NTP) (Roche, Basel, Switzerland) at 37°C for 0.5 h, and then cultured in DMEM supplemented with 2 mM CaCl2 at 37°C for 2 h for membrane resealing (Håkelien et al., 2002). After resealing, the remaining cells were cultured in ES medium (Vejlsted et al., 2005; DMEM/F12 (Gibco, Grand Island, NY), 5% FCS, 10% KnockOut Serum Replacement (Gibco), 1% penicillin/streptomycin, 1 mM glutamine, 0.3 μM nucleotides, 143 μM 2-mercaptoethanol, MEM NEAA, and 10 ng/mL LIF (Chemicon, Tumecula, CA) for 2 to 12 days.
The treated cells from each extract batch reached 90% confluence on Day 2, when they were split on two coverslips placed in a four-well dish and cultured for further 10 days. The culture medium was changed every 2 days. Cell clusters formed on Days 5 to 6 after extract treatment, and these clusters became colonies on Days 7 to 8 and grew even bigger on Day 12. The colonies formed on the coverslips in each well were counted on Day 7. For each batch of extract, four to five independent treatments were performed, and the colony numbers were counted from 12 to 16 coverslips. Our preliminary experiment showed that the extract treated cells but without digitonin treatment could not induce colony formation in treated cells.
Using colony cells for HMC
Cell colonies from each extract batch cultured for 7 to 8 days after extract treatment were collected by mouth pipette and trypsinized for 10 min, and then separated into single cells by pipetting in TCM-199 with 20% cattle serum (CS; Danish Veterinary Institute, DTU). These single cells were then directly used for HMC. Nontreated cells cultured in DMEM with 10% FCS were used for control group. For testing the effect of culture medium on cloning, nontreated cells cultured in both DMEM with 10% FCS and ES medium were used for cloning. To synchronize the donor cells for HMC, culture medium of all the colony cells and nontreated cells grown in DMEM and ES medium were not changed for 2 to 3 days.
HMC was performed as described before (Du et al., 2005; Liu et al., 2011). Briefly, matured cumulus–oocyte complexes (COCs) were freed from cumulus cells with 1 mg/mL hyaluronidase (Day 0). After partial digestion of zona pellucida with 3.3 mg/mL pronase in TCM-199 with 33% CS, oriented bisection of oocytes was performed manually with microblade (AB technology, Pullman, WA) in TCM-199 with 2% CS drop supplemented with 2.5 mg/mL cytochalasin B under a stereomicroscope. Each cytoplast without polar body attached with a single treated or nontreated cell was fused in fusion medium [0.3 M mannitol, 0.1 mM MgSO4, and 0.01% (w/v) PVA] in a fusion chamber (BTX microslide 0.5-mm fusion chamber, model 450; BTX, San Diego, CA) with single direct current (DC) impulse of 2.0 kV/cm for 9 μsec. One hour later, each cytoplast–somatic cell pair was fused with another cytoplast in activation medium (fusion medium with 0.1 mM CaCl2) by a single DC pulse of 0.86 kV/cm for 80 μsec. After incubation in porcine zygote medium 3 (PZM-3) (Yoshioka et al., 2002) supplemented with 5 μg/mL cytochalasin B, 10 μg/mL cycloheximide for 4 h, the reconstructed embryos were cultured individually in WOWs (Vajta et al., 2000) made in four-well dishes filled with PZM-3 medium. For capacity reasons, one or two groups of extract-treated cells and one control group were used on each cloning day. Accordingly, each group of treated cells had their own control, but the number of replicates varies between groups.
Cleavage and blastocyst rates as well as total cell number per blastocyst were checked on Days 2 and 6, respectively. For assessing the total cell number, the blastocysts at Day 6 from each group were stained with 10 μg/mL Hoechst 33342 for 20 min and mounted on a slide in 5 μL of glycerol. The number of cells was then counted under a fluorescence microscope.
Analysis of pluripotency markers expression in extract-treated cell
Treated cells were used for analysis of expression of pluripotent markers 8 days after extract treatment, representing the cells at the colony stage and when the colony cells were used for cloning. Expression of pluripotency markers (OCT4, CDH, SOX2, and NANOG) and reference gene (beta-ACTIN) in extract-treated cells were examined by reverse transcription (RT) PCR. RNA of cells was isolated by mirVana RNA isolation kit (Ambion, Austin, TX). DNase treatment was used to avoid genomic contamination. Reverse transcription was performed with 200 ng RNA using M-MuLV reverse transcriptase (Finnzymes, Pittsburgh, PA), Oligo (dT)12-18 Primer (Invitrogen, Carlsbad, CA), DNTP (Invitrogen), and RNase inhibitor (Fermentas, Germany). PCR amplification was carried out on 1 μL of the RT product per 20 μL of PCR reaction mixture containing 9 μL water, 2 μL 10x PCR buffer, 2 μL 2.5 mM MgCl2, 2 μL DNTP, 2 μL sense primer, 2 μL antisense primer, and 0.14 μL Taq gold pol. Sequences of the primers are listed in Table 1. RT-PCR conditions were 95°C for 12 min, 35 cycles of 95°C for 15 sec, 60°C for 30 sec, 72°C for 45 sec. The PCR products were detected on 1.5% agarose gels under ultraviolet light.
Statistical analysis
Cleavage and blastocyst rates were analyzed with a chi-square test, and colony number and blastocyst cell number were analyzed by Duncan's multiple comparison (SAS version 9.2). A probability of p<0.05 was considered to be statistically significant.
Results
Colony formation in different batches of extract-treated cells
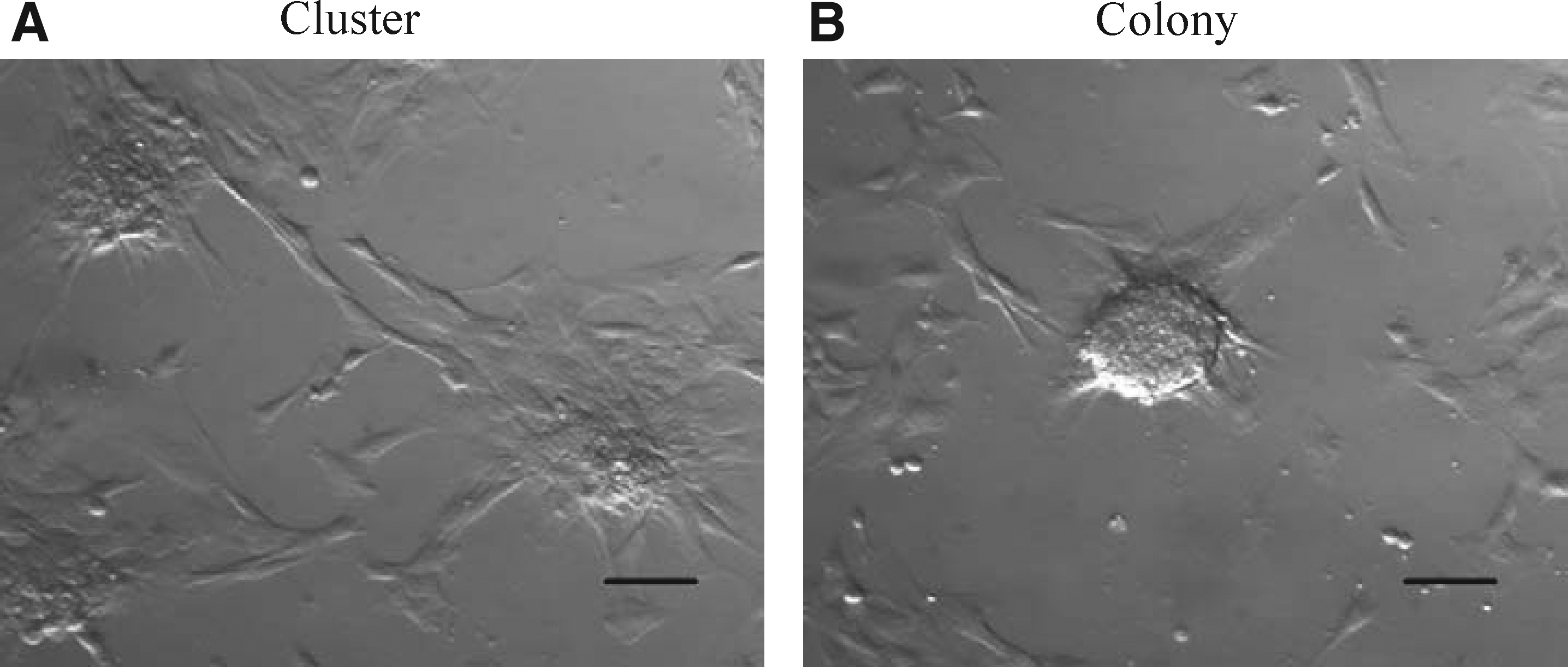
Various cell aggregates (clusters) began to form on Days 4 to 5 after extract treatment (Fig. 1A). On Day 7 after extract treatment, the cell clusters formed well-defined colony structures (Fig. 1B). The colony formation was observed in the treated cells from all three batches, but Batch 2 extract-induced formation of a higher number of colonies than the other two batches (p<0.05). No colony formation was detected after use of both nontreated cell and cells with digitonin treatment alone grown in DMEM and ES medium. The protein concentration in the extracts and the number of colonies in extract-treated cells from all three batches are shown in Table 2.
(
Different superscripts in the same column indicate significant differences: a versus b (p<0.05).
Development of cloned embryos with colony cells from different batches of extract
A total of 564 reconstructed embryos from at least three replicates for each group were produced. No significant differences were observed in the cleavage rates of cloned embryos and total cell number of cloned blastocysts reconstructed with colony cells from the different batches of extract and control group (Table 3). Interestingly, blastocyst rate after using Batch 2 colony cells was significantly higher than after using Batch 3 colony cells or nontreated cells (56 vs. 43% and 45%, p<0.05). As the blastocyst rate of embryos cloned with Batch 2-treated cells was higher than for the other two batches and the control group, more replicates were performed only with Batch 2 and control cells.
Values with different superscripts in the same column are statistically different (p<0.05).
Furthermore, we investigated possible influence of donor cells' culture media on cloning efficiency (DMEM for control cells and ES medium for Xenopus extract-treated cell donors). However, no significant differences in cleavage rate, blastocyst rate, and blastocyst's cell numbers were observed after using nontreated cells grown in DMEM and ES medium (Table 4).
No significant difference: p>0.05.
Pluripotency markers expression in colony cells after extract treatment
Beside colony formation, gene expression of pluripotency markers (OCT4, CDH, SOX2, and NANOG) in colony cells on Day 8, which is the stage when donor cells were used for SCNT, was evaluated by RT-PCR (Fig. 2). OCT4 expression was detected in colony cells treated with all three batches. In parallel control groups, OCT4 was not detected in the nontreated cells grown in DMEM and/or ES medium, nor in the cells permeabilized with digitonin. CDH transcripts were detected only in colony cells from Batch 2 and 3 extract, but not in Batch 1 extract. No CDH expression was detected in the three control groups. SOX2 expression was detected in colony cells from all three batches of extract, which is similar to OCT4 expression, and no expression was detected in control groups. NANOG expression was found both in colony cells and three control groups.
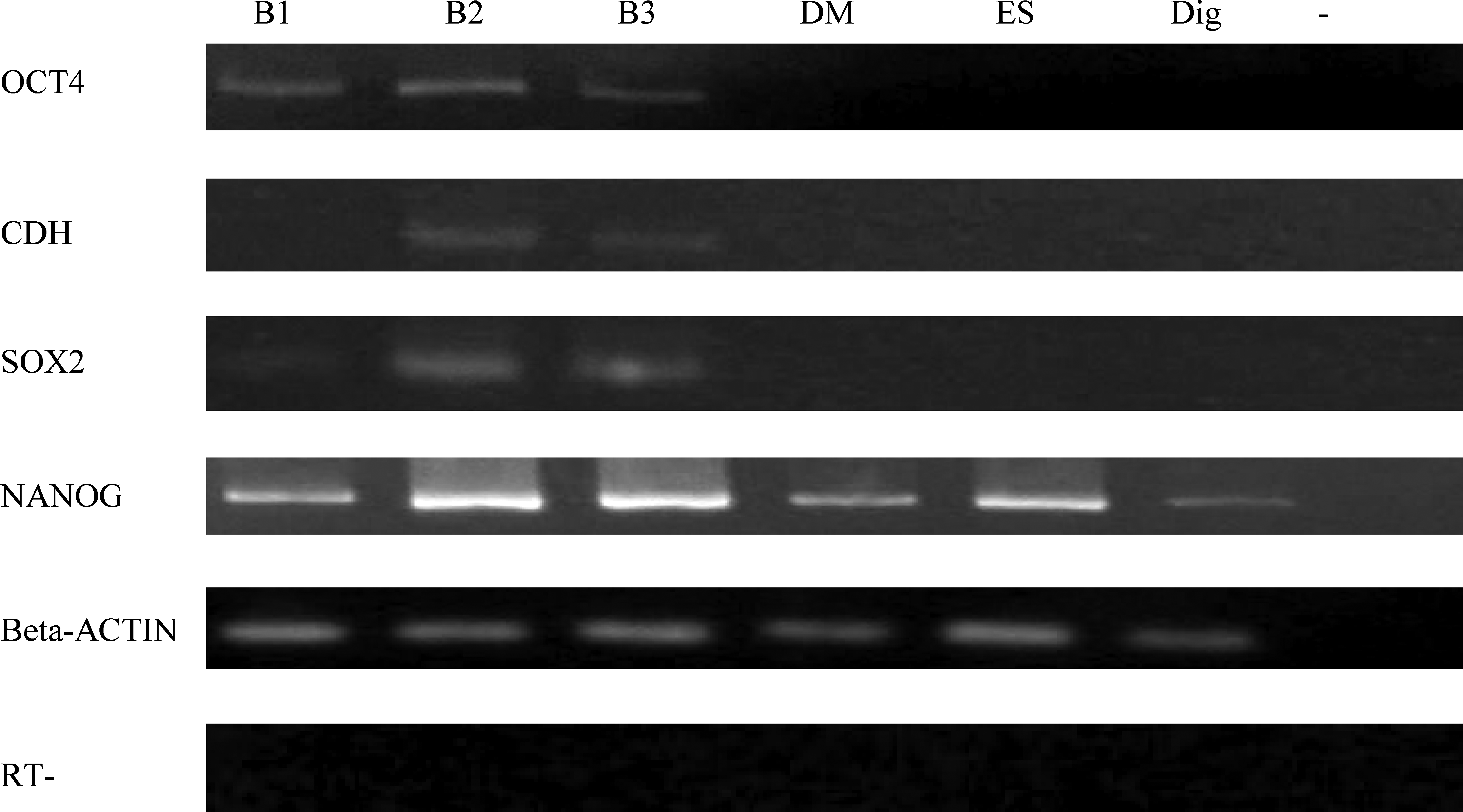
Expression of pluripotency markers (OCT4, CDH, SOX2, and NANOG) in colony cells on Day 8 from different batches of extract (B1, 2, and 3=Batch 1, 2, and 3). Beta-ACTIN was used as a control. DM, nontreated cells grown in DMEM; ES, nontreated cells grown in ES medium; Dig, digitonin-treated cells grown in DMEM; −, H2O. RT, isolated RNA without reverse transcription to check genomic DNA contamination.
Discussion
Extracts from eggs of Xenopus laevis frogs are models traditionally used for studies of chromatin (Laskey et al., 1977) and nuclear assembly (Lohka and Masui, 1983) as well as DNA replication (Blow and Laskey, 1986). Recently it has been shown that pretreatment with Xenopus extract can induce reprogramming of somatic cells (Alberio et al., 2005; Hansis et al., 2004; Miyamoto et al., 2007, 2008) and increase cloning efficiency seen as higher live birth rate (Rathbone et al., 2010). However, the knowledge of active substances in such extracts, affecting cellular morphology and gene expression, are still limited, and at present there is no effective method to characterize the activity of extract. Indeed, in a previous study we have shown that pretreatment of porcine donor cells with Xenopus egg extract increased SCNT blastocyst formation (Liu et al., 2011). In the present study, we demonstrate the effect of the frog extract on reprogramming porcine somatic cells, resulting in up to 20% higher blastocyst formation, which was also related to the colony formation induced by the extract. On the other hand, we also found blastocyst formation of cloned embryos varied when using donor cells pretreated with different batches of Xenopus egg extract, which indicated that the activity of extracts is variable from batch to batch.
In our experiments, the growth pattern was changed in extract-treated cells during cell culture, as cell clusters and colonies formed, and as the number of colonies formed differed when different batches of extract were used. Similar morphological changes of extract-treated cells had also been observed in previous studies (Hansis et al., 2004; Miyamoto et al., 2007). Considering possible physical extract characteristics (protein concentration), we did not reveal any correlation between colony formation and extract type, as Batch 1 and 3 with low and high protein concentrations, respectively, both induced a low number of colonies in treated cells. These few results indicated that the protein concentration may not sufficiently characterize the quality and activity of the extracts. Importantly, our results show a positive correlation between the ability of a batch of egg extract to induce colonies and increased blastocyst formation when these colony cells are used in production of porcine somatic cell nuclear transfer (SCNT) embryos, as better blastocyst formation was observed when using cells treated with the batch of extract showing the higher colony formation. This result suggested the egg extract, which induced higher colony formation, may have better biochemical activity for cloning.
One possible reason could be the reprogrammed cells induced by Xenopus egg extract may be more accessible to full-term after cloning. Our results show that some pluripotent markers were expressed in the colony cells that were induced by extract, but that the cells did not reach to full pluripotent stage as NANOG expression was detected in both extract-treated and nontreated cells. When using these colony cells as donors for cloning, another round of nuclear reprogramming is induced by oocyte cytoplasm, which may result in full reprogramming in donor cells for cloning. Moreover, OCT4 and SOX2 expression was detected in the colony cells from all three batches extracted in this study, but CDH expression was only detected in Batch 2 and 3, which indicates the expression of OCT4 and SOX2 is enough to support colony formation.
Another possible reason could be that the epigenome (Miyamoto et al., 2007; Rathbone et al., 2010) as well as the chromatin morphology (Pyne, 2001), are structurally loosened in extract, which should be ready for attachment of reprogramming factors, to perform “normal” remodeling in extract and oocyte cytoplasm, just like the mature oocytes affect sperm. On the other hand, the cells from the extract inducing the higher colony formation may undergo a better remodeling process than other extract-treated cells with lower colony formation, as the cloning efficiency was increased.
In conclusion, our study demonstrates that Xenopus egg extract can induce reprogramming in porcine somatic cells, and that assessment of colony formation may be a method to select reprogramming ability of extract for pretreatment of fibroblasts to increase cloning efficiency. Also, our study may open up for new possibilities to reveal the active substance(s) in extract that induce reprogramming of donor cells and that is beneficial for SCNT.
Footnotes
Acknowledgments
The authors thank Anette M. Pedersen, Janne Adamsen, Klaus Villemoes, Ruth Kristensen, Annette K. Nielsen, and Hanne Jørgensen for excellent technical assistance. The work was supported financially by grants from the “Nutriomics” project (Danish Agency for Science, Technology and Innovation, 2101-06-0034), the “Pigs & Health” project (Danish National Advanced Technology Foundation, 013-2006-2), and Carlsberg foundation 2010-01-0452.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.