
Abstract
Abstract
The mechanisms of nuclear reprogramming following somatic cell nuclear transfer (SCNT) to enucleated oocytes or factor-based reprogramming are poorly understood. In this study global transcriptional analysis was performed on a number of different rhesus monkey (Macaca mulatta) cell and tissue samples, including rhesus-induced pluripotent stem cells (IPSCs) and rhesus SCNT-derived embryonic stem cells (SCNT-ESCs). Global transcriptional cluster analysis and stem cell-specific gene expression analysis both suggested that the oocyte-reprogrammed SCNT-ESCs were transcriptionally closer to the control fertilized ESCs than IPSCs. These results, combined with previous epigenetic analysis studies in the mouse, reinforce the hypothesis that oocyte-reprogrammed cell nuclei are more completely reprogrammed to an ESC state than IPSCs. Transcriptional analysis of rhesus oocytes detected over 500 ESC-specific genes, including OCT3/4, NR5A2, and DNMT3B. These results, combined with previously published reprogramming research, were used as the basis for a general model to explain the mechanisms of nuclear reprogramming.
Introduction
Materials and Methods
Rhesus macaque cell lines and tissues analyzed
Affymetrix CEL files for the rhesus macaque cell lines and tissues analyzed in this study were obtained from the Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo). Previous research has demonstrated that rhesus macaque ESCs (Mitalipov et al., 2006), SCNT-ESCs (Byrne et al., 2007) and IPSCs (Liu et al., 2008) express stem cell markers, possess a normal karyotype, and can differentiate into teratomas containing representatives of all three germ layers (Supplementary Table 1; Supplementary Data are available online at www.liebertonline.com/cell). At least two replicates for each cell line or tissue type were included in each comparison analysis. Each CEL file was generated through analysis of cell line or tissue total RNA hybridized to the Rhesus Affymetrix GeneChip array (Byrne et al., 2007; Duan et al., 2010; Lee et al., 2008; Liu et al., 2008). The following CEL files were analyzed and compared in this study: GSM187389 (fibroblast line A replicate 1), GSM187390 (fibroblast line A replicate 2), GSM187392 (ESC line A replicate 1, GSM187393 (ESC line A replicate 2), GSM187395 (ESC line B replicate 1), GSM187396 (ESC line B replicate 2), GSM187398 (SCNT-ESC line A replicate 1), GSM187399 (SCNT-ESC line A replicate 2), GSM187401(SCNT-ESC line B replicate 1), and GSM187402 (SCNT-ESC line B replicate 2) from Series GSE7748 (Byrne et al., 2007), GSM329248 (fibroblast line B replicate 1), GSM329249 (fibroblast line B replicate 2), GSM329251 (ESC line C replicate 1), GSM329252 (ESC line C replicate 2), GSM329254 (IPSC replicate 1), and GSM329255 (IPSC replicate 2) from Series GSE13146 (Liu et al., 2008), GSM241366 (cortex replicate 1), GSM241367 (cortex replicate 2), GSM241374 (thymus replicate 1), GSM241375 (thymus replicate 2), GSM241370 (pancreas replicate 1), and GSM241371 (pancreas replicate 2) from Series GSE9531 (Duan et al., 2010), and GSM300529 (metaphase II oocytes replicate 1) and GSM300530 (metaphase II oocytes replicate 2) from Series GSE11895 (Lee et al., 2008). Please note that the rhesus IPSCs were generated through retroviral expression of the Yamanaka reprogramming factors (OCT3/4, SOX2, KLF4, and cMYC) (Liu et al., 2008).
Data analysis
Each CEL file was uploaded to GeneSifter (VisX Labs, Seattle, WA) using the Advanced Upload Method and normalized using the Affymetrix Microarray Analysis Suite (MAS) 5.0 algorithm. Cluster analysis between groups of samples was performed through GeneSifter Project Analysis using analysis of variance (ANOVA) statistical analysis (p<0.01, threshold > 20, Manhattan distance, ward linkage, and gene row centering). GeneSifter pairwise analysis between samples was performed using all mean normalization and t-test statistical analysis (p<0.01). For each pairwise analysis at least two replicates from each cell line or tissue type was compared to a baseline that consisted of pooled rhesus monkey dermal fibroblasts from two different experiments (GSM187389, GSM187390, GSM329248, GSM329249). Probe sets were considered to be significantly upregulated (compared to the pooled fibroblast baseline) when the p-value was <0.01 and fold change was equal or greater than 3. When duplicate probe sets or genes were identified the duplicates with the lower fold change were removed. Gene ontology analysis for biological processes was performed in GeneSifter on the significantly upregulated probe sets.
Statistical analysis
The ANOVA statistical analysis and t-test statistical analysis were performed in GeneSifter with p-values set at less than 0.01. The Pearson product–moment correlation coefficient formula was used to measure the correlation between the upregulated expression levels (relative to the baseline pooled fibroblasts) of the principal ESC-specific genes in the various cell lines and tissue types compared with the pooled ESCs.
Results
Global cluster analysis
Cluster analysis of the rhesus monkey fibroblasts and rhesus monkey ESCs from different experiments (GSE7748 and GSE13146) demonstrated that each cell line clustered according to its cell type (Fig. 1A). This highlighted that potential global transcriptional differences from intrinsic factors, such as cell line to cell line variability and extrinsic factors such as possible experimentally induced differences, were significantly lower than the global transcriptional similarities due to cell type. Nevertheless, in order to further minimize any intrinsic and extrinsic variability, multiple replicates of each of the control samples (fibroblasts and ESCs) were respectively pooled from the two different experiments (GSE7748 and GSE13146) for all subsequent analyses (Fig. 1A). Next, cluster analysis was performed on the pooled fibroblasts, pooled ESCs, and on the reprogrammed cell lines, which included the rhesus monkey SCNT-ESCs and rhesus monkey IPSCs. This analysis demonstrated that the SCNT-ESCs clustered more tightly with the control ESCs than the IPSCs (Fig. 1B) providing the first indication that the oocyte-reprogrammed rhesus SCNT-ESCs may be more effectively reprogrammed toward an ESC transcriptional state than the factor-reprogrammed IPSCs. Previous epigenetic analysis of mouse SCNT-ESCs and mouse IPSCs using comprehensive high-throughput array-based relative methylation (CHARM) has demonstrated a similar clustering pattern between mouse ESCs and syngeneic mouse SCNT-ESCs, which cluster together, and syngeneic mouse IPSCs, which cluster separately (Kim et al., 2010). The preliminary clustering result reported here supports the hypothesis that primate (rhesus monkey and presumably human) ESCs and SCNT-ESCs will tend to cluster more closely than primate IPSCs following either epigenetic and/or transcriptional analysis. Although epigenetic analysis was not performed in this study, it is probable that any statistically significant differences in gene transcription reflect differences at the epigenetic level.

Global cluster and gene ontology analysis. (
It should be noted that gene ontology analysis was performed in GeneSifter and did not reveal any major differences in the categories of upregulated genes (when compared to the pooled fibroblasts) across ESCs (Fig. 1C), SCNT-ESCs, IPSCs, cortex tissue, thymus tissue, and pancreatic tissue (data not shown).
Analysis of ESC-specific gene expression in SCNT-ESCs and IPSCs
In order to further dissect the effectiveness of the SCNT and IPSC reprogramming methodologies, it was necessary to identify a list of genes specifically expressed in ESCs. Pairwise comparison analysis was performed between the pooled fibroblasts and the pooled ESCs, and 1669 stem cell specific genes were identified that demonstrated an equal or greater than threefold significantly (p<0.01) increased level of gene expression in the pooled ESCs compared with the pooled fibroblasts (Supplementary Table 2). These stem cell specific genes were sorted by fold change and the top 40 genes were identified as the principle ESC specific genes (Table 1). Pairwise comparison analysis—against the baseline pooled fibroblasts—was performed on a control ESC line (ESB line B)—an ESC line that was not used in the ESC pooled sample—and on the SCNT-ESCs and IPSCs (Supplementary Table 3). Next, the principle ESC-specific genes were identified from each of the pairwise comparisons and the relative increased fold changes for each gene—compared to the baseline fibroblasts—was recorded (Table 1).
These genes are expressed in IPSCs at less than 70% of their expression level in the pooled ESC controls.
ESCs, embryonic stem cells; SCNT, somatic cell nuclear transfer; IPSCs, induced pluripotent stem cells.
In order to establish the extent of reprogramming of these principle ESC-specific genes the Pearson correlation coefficient—with the principle ESC-specific genes in the pooled ESCs—was measured across various cell lines and tissue types. As a positive control the ESCs (ESC line B) demonstrated, as expected, a very high correlation coefficient of 0.94 (Table 1), whereas, as negative controls, various somatic tissues demonstrated, as expected, very low correlation coefficients, which included 0.003 for cortex tissue, 0.098 for thymus tissue, and 0.066 for pancreatic tissue (Supplemental Table 3). Interestingly, the SCNT-ESCs demonstrated a correlation coefficient of 0.91, whereas the IPSCs demonstrated a lower correlation coefficient of 0.81, providing additional evidence to support the hypothesis that SCNT-ESC are, in general, closer to the ESC transcriptional state than IPSCs, although confirmation with larger data sets is required.
When the level of principle ESC-specific gene expression was compared across the different pluripotent stem cell lines, it became apparent that almost half (19/40) of the IPSC principle ESC-specific genes were not fully reprogrammed and had a gene expression level that was less than 70% of that observed in the pooled ESCs (Table 1 and Fig. 2). The SCNT-ESCs had only three principle ESC-specific genes expressed at less than 70% of the pooled ESC level, which was the same number as observed in the control ESCs (Fig. 2).

Relative gene expression of principle ESC-specific genes in reprogrammed cell lines. Relative gene expression of the principle rhesus monkey ESC-specific genes was examined in control ESCs (line B), SCNT-ESCs, and IPSCs. The orange line under the x-axis indicates the level at which gene expression was 70% (0.7) of that observed in the pooled ESCs. Genes with an asterisk were also detected at significant levels in metaphase II rhesus monkey oocytes.
Detection of ESC-specific genes in rhesus monkey oocytes
The global cluster analysis and ESC-specific gene expression analysis both suggested that the SCNT-ESCs had more faithfully reprogrammed to the ESC transcriptional state than the IPSCs. One possible hypothesis to explain this difference was that the rhesus monkey metaphase II oocytes used in SCNT possess a large number of ESC-specific factors and these factors significantly augmented the reprogramming process. In order to test this hypothesis pairwise comparison analysis was performed with rhesus monkey metaphase II oocytes and the pooled fibroblasts, and a list of significantly (p<0.01) “upregulated” genes with equal or greater than threefold change in gene detection/expression was identified (Supplementary Table 4). Next, this list of oocyte genes was screened for ESC-specific genes and 593 ESC-specific genes were identified (Supplemental Table 4), which included 20 of the principle ESC-specific genes (Table 2). This data demonstrates that rhesus monkey metaphase II oocytes possess significant amounts of maternally derived transcripts for a large number of ESC-specific genes.
Discussion
In this study global cluster analysis and ESC-specific gene expression analysis was performed on both rhesus monkey SCNT-ESCs and rhesus monkey IPSCs and both the cluster analysis and ESC-specific gene expression analysis suggested that the SCNT-ESCs had more faithfully reprogrammed to the ESC transcriptional state than the IPSCs. Although this study represents only a preliminary analysis of oocyte-based and factor-based nuclear reprogramming, the extremely limited number of rhesus monkey SCNT-ESC lines (Byrne et al., 2007) and rhesus monkey IPSCs (Liu et al., 2008) makes a larger comparison study currently not possible. In addition, although human SCNT embryos have been generated (French et al., 2008; Stojkovic et al., 2005) no human SCNT-ESC lines have been derived to date, which makes this rhesus monkey transcriptional analysis the only method currently available for comparing oocyte-based reprogramming and factor-based reprogramming in the primate. Future transcriptional and epigenetic analysis of a larger number of isogenic rhesus monkey samples, in addition to deeper mechanistic reprogramming research, will provide definitive evidence either for or against the conclusions and model proposed here. The differentially expressed gene lists identified in this study, for example the principle ES-specific genes or the oocyte specific genes, and their relative fold changes, are derived from microarray comparison analysis, as described in the methods, and a more detailed examination of the relative expression of these genes using quantitative RT-PCR (or similarly sensitive transcriptional analysis technologies) may reveal significant differences with the global microarray-derived gene expression fold changes reported here. Both the rhesus SCNT-ESCs and IPSCs were transcriptionally analyzed at an early passage (passage 10 or less; Hongkui Deng, personal communication). It has been demonstrated that transcriptional differences between lower passage (passage 10 or less) human IPSCs and human ESCs will significantly reduce over extended culture; for example, when STEMCCA-IPSCs where expanded to over 20 passages (Chin et al., 2010). Therefore, it is possible that the transcriptional differences observed in this preliminary study may significantly decrease with higher passage IPSCs, as has been observed for human IPSC lines (Chin et al., 2010), an important consideration for future analysis.
The primate oocyte is able to reprogram the rhesus somatic cell nucleus to pluripotency in less than a week (Byrne et al., 2007) with this preliminary study suggesting that over 92% of the principle ESC-specific genes are fully reprogrammed (Fig. 2), whereas the factor-based reprogramming methodology takes about a month (Liu et al., 2008) and nearly half (48%) of the principle ESC-specific genes were not fully reprogrammed (Fig. 2). Transcriptional analysis of the rhesus monkey oocytes demonstrated that these oocytes possess significant amounts of maternal transcripts for over 500 ESC-specific genes, including half of the principle ESC-specific genes and such key stem cell regulators as OCT3/4 (POU5F1) and NR5A2, the latter of which has been demonstrated to be able to replace OCT3/4 in reprogramming experiments (Heng et al., 2010). Based on this information we can ascertain that immediately following SCNT to a primate oocyte, the somatic nucleus is placed into an environment with over 500 ESC-specific genes/factors, including high levels of the OCT3/4 transcription factor and the epigenetic modifier DNMT3B, a key component in resetting the DNA methylation code, while following transduction with defined factors the infected cell builds up expression of only four genes, OCT3/4, SOX2, KLF4, and cMYC following viral transduction, integration, and expression from the genome (Fig. 3). In addition to the large number of ESC-specific factors residing in the oocyte, the oocyte also contains a large amount of cytoplasm that is capable of quickly diluting the somatic transcription factors and somatic microRNAs following nuclear transfer, whereas the transduced somatic cell only contains a small cytoplasmic space that maintains a high concentration of the somatic determinants. It should also be noted that although the factor-based reprogramming is driven by the static exogenous expression of four factors, the gene expression environment following SCNT is developmentally dynamic, gradually changing over embryonic development, as determined initially by the maternal RNA, and by the blastocyst-stage involving expression of the rest of the major rhesus stemness factors not initially contained in the oocyte, such as Nanog (Harvey et al., 2009).
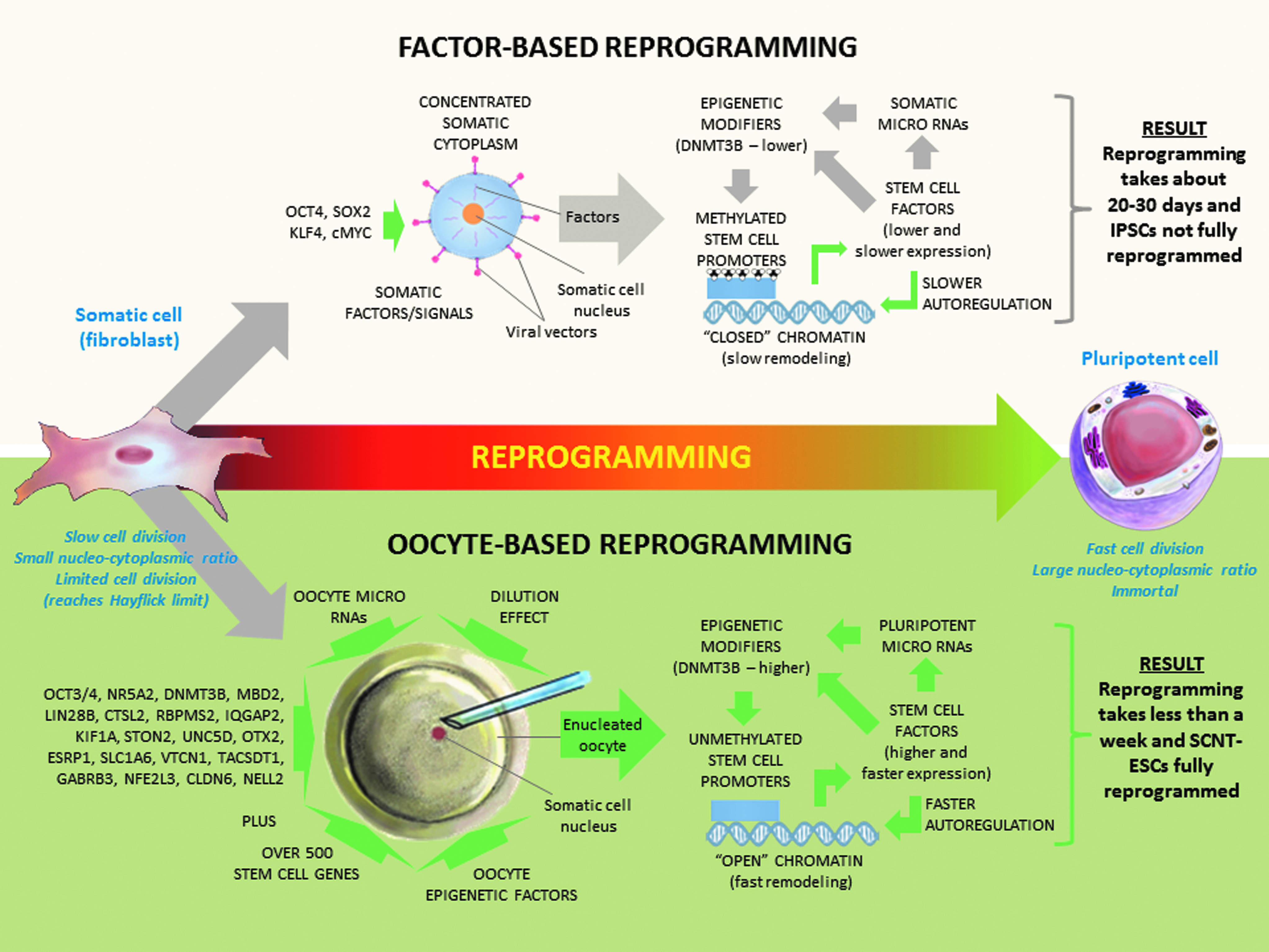
General model to explain the mechanisms of nuclear reprogramming. Generalized model to explain the mechanisms of nuclear reprogramming for defined factor-based and oocyte-based reprogramming.
The preliminary data obtained from this study can now be combined with recently published research from the nuclear reprogramming field (Anokye-Danso et al., 2011; Kim et al., 2010) to provide a generalized three stage model explaining the nuclear reprogramming mechanism (Fig. 3). This model includes chromatin remodeling, autoregulatory reprogramming, and downstream effects (which includes the microRNA environment, epigenetic modifiers, and epigenetic code). These three stages form a reprogramming cycle that progress continuously in parallel, driven by reprogramming factors from the oocyte or exogenously delivered into the cell, until a stable epigenetic pluripotent state is obtained.
Chromatin remodeling
The first stage involves nuclear remodeling of the chromatin following either transfer to the oocyte cytoplasmic environment or following transduction with the defined reprogramming factors. In factor-based reprogramming, exogenous OCT3/4 and SOX2 first bind genes that encode chromatin-remodeling factors, such as SMARCAD1, MYST3, JMJD1A, and JMJD2C (Loh et al., 2006, 2007) and the gradual increase in the expression of these factors leads to a general conversion of the more condensed somatic chromatin toward a more open embryonic state (Fig. 3). Following nuclear transfer to the primate oocyte, a rapid breakdown of the somatic cell nuclear membrane is observed followed by a rapid chromatin remodeling (Mitalipov et al., 2007). This rapid remodeling over the embryonic cleavage divisions results in a quick conversion of somatic chromatin structures into initially oocyte-based and then embryonic-based chromatin structures as the nuclear transfer embryo proceeds through development (Fig. 3).
Autoregulatory reprogramming
Once the chromatin has remodeled to an accessible state, the promoters of the stem cell genes can be accessed and reprogrammed by those same stem cell factors in a form of autoregulatory reprogramming. In factor-based reprogramming, exogenous OCT3/4 and SOX2, once expressed, co-occupy and activate the endogenous OCT3/4 and NANOG genes (Kuroda et al., 2005; Okumura-Nakanishi et al., 2005). OCT3/4, SOX2, and NANOG then bind together both at their own promoters and at the promoters of target genes and form an interconnected autoregulatory loop (Boyer et al., 2005; Loh et al., 2006). This autoregulatory reprogramming is initially driven by only a small number of factors, but as a broader spectrum of endogenous stem cell genes become activated, these augment and gradually speed up the process in both a cell division-dependent and Nanog expression-dependent manner (Hanna et al., 2009). In oocyte-based reprogramming, this study has identified that over 500 stem cell specific genes/factors are already present in the primate oocyte, which may drastically speed up the establishment of a broad interconnected autoregulatory loop, and as the nuclear transfer embryo proceeds through embryonic development toward the blastocyst, this broad interconnected autoregulatory loop rapidly incorporates expression of all the key stem cell marker genes (Byrne et al., 2007).
Downstream effects
As the autoregulatory reprogramming process is occurring, the exogenous and reprogrammed endogenous factors exert their complex downstream effects through induction and regulation of both microRNAs and epigenetic modifiers that can, in turn, further interact with transcription factor expression and each other, and reset the epigenetic code back into a pluripotent epigenetic state. This is a complex interconnected process that, as a field, is being gradually refined for better understanding. However, certain aspects of these downstream reprogramming effects are now understood. For example, recent studies have found that both OCT3/4 and SOX2 are crucial for expressing mir-302, a key pluripotency associated microRNA, in human ESCs (Card et al., 2008; Marson et al., 2008). Mir-302 in turn suppresses AOF2, a histone demethylase, and results in a significant decrease in DNA methyl transferase 1 (DNMT1). Lower levels of DNMT1 cause global demethylation that is augmented by MECP1/2 suppression (Lin et al., 2011). However, low DNMT1 expression is a slow passive demethylation process, highlighting the slow (multiweek) demethylation observed following factor-transduction (Mikkelsen et al., 2008), whereas research has suggested that the mammalian oocyte possesses an rapid (and as-of-yet unknown) active demethylation ability (Santos et al., 2002). Cell fusion studies by Helen Blau and colleagues have identified in ESCs an activation-induced cytidine deaminase (AID) as being essential for active OCT3/4 and NANOG promoter demethylation (Bhutani et al., 2010). However, the only cytidine deaminase differentially expressed from this transcriptional analysis study was CDA (cytidine deaminase) and this gene demonstrated a greater than twofold higher expression in the pooled fibroblasts compared to the rhesus oocytes (p<0.05), suggesting that oocytes may use a different mechanism for their active demethylation. It is not known how exactly the microRNA environment of IPSCs compares to SCNT-ESCs; however, research examining control fertilized embryos and SCNT embryos found them to be indistinguishable in their microRNA profiles (Ding et al., 2009). This study has detected significant DNMT3B mRNA in primate oocytes (Table 2), suggesting that this epigenetic modifier may play a key role in resetting the DNA methylome toward a pluripotent epigenetic state. It was also observed that there was lower DNMT3B mRNA levels in the rhesus monkey IPSCs analyzed compared to the rhesus SCNT-ESCs (Table 1), suggesting that expression of this key epigenetic modifier may maintain a higher level of expression throughout the SCNT process compared with the factor-based reprogramming. However, it should be noted that as this is a transcriptional analysis, further validation will be required to confirm that DNMT3B is differentially expressed at the protein level.
It is interesting to note that while the factor-based reprogramming cycle has traditionally been driven by the four transcription factors (OCT3/4, SOX2, KLF4, and cMYC) the reprogramming cycle could theoretically be maintained from any of the three major nodes: transcription factors, microRNAs, or epigenetic modifiers. A recent study has demonstrated that the microRNAs 302 and 367 alone are capable of driving the reprogramming of human somatic cells to a pluripotent state without requiring exogenous expression of transcription factors (Anokye-Danso et al., 2011). It appears that miRNA302/367 repress a large number of mRNA targets that, in turn, regulate chromatin remodeling and cell proliferation, and mi367 appears to be an essential component of the induced reprogramming cycle (Anokye-Danso et al., 2011; Krek et al., 2005). Also, Sheng Ding and colleagues have recently demonstrated that two small molecules AMI-5 (a protein arginine methyltransferase inhibitor) and A-83-01 (a TGF-β inhibitor) are capable or reducing the required number of reprogramming factors to just OCT3/4 (Yuan et al., 2011). Eventually we may discover a combination of chemical or biological epigenetic modifiers and/or small molecules capable of inducing pluripotency without requiring the exogenous expression of either transcription factors or microRNAs.
Although the isogenic pluripotent stem cell field has been considered to have the potential to have an immense impact on future human autologous cellular therapies (Byrne, 2008), recent murine work has demonstrated that a number of factor-reprogrammed IPSC lines aberrantly reprogrammed genes (Hormad1 and Zg16) that resulted in an induced immune rejection, even following transplantation into syngeneic inbred mice (Zhao et al., 2011) a potentially major barrier for the usage of human factor-reprogrammed cells in cellular therapies. There are several questions that should be addressed before we conclude that human IPSC-derived cells will be rejected following patient-specific autologous cellular therapies: (1) are Hormad1 and Zg16 already highly expressed in mouse embryonic fibroblasts (MEFs) compared to mouse adult tail tip fibroblasts and/or other adult cell populations? If so, it is possible the immunogenicity issues observed in the syngeneic MEF-IPSC-teratomas are simply due to faulty reprogramming of these two genes from MEFs and these problems would not be observed with reprogrammed adult cells; (2) does the teratoma generate a subset of cells that are exacerbating the immune response? For example, the teratoma may contain embryonic, fetal, and/or rapidly dividing cell types that may “sensitize” the immune response because of their rarity in adult mice. Indeed, careful examination of the Zhao et al. (2011) article does reveal some CD3-expressing cell infiltration into an ESC-derived teratoma in a syngeneic host, providing a degree of support for this hypothesis; and (3) are only a small fraction of IPSC lines nonimmunogenic, similar to the small fraction of mouse IPSC lines that can be used to generate mice through tetraploid complementation (Boland et al., 2009; Zhao et al., 2009). The six IPSC lines examined by Zhao et al. (2011) may have been an insufficient number to identify these rare fully reprogrammed nonimmunogenic IPSC lines. Despite the unresolved nature of these questions, it remains possible that patient-specific IPSC-derivatives generated with contemporary technology will indeed be rejected by the immune system. This immune rejection of reprogrammed cells has not been observed following autologous transfer of SCNT oocyte-reprogrammed murine cells (Tabar et al., 2008), suggesting that the oocyte possesses key factors necessary to fully reprogram somatic cells into an immune-competent pluripotent state. The results presented here support the hypothesis that the primate oocyte contains factors that tend to induce a more complete reprogramming to an ESC state (Fig. 1B and Fig. 2), similar to a previous report in the mouse (Kim et al., 2010). However, oocyte-based reprogramming in the primate requires a large number of fresh metaphase II oocytes in order to generate pluripotent stem cells (Byrne et al., 2007) a significant limitation in extending these studies to the human (Byrne, 2008). These recent developments and pragmatic considerations suggest that a prudent approach toward future patient-specific cellular therapies would include detailed comparison analysis of oocyte-based and factor-based reprogramming to determine the key oocyte-based factors that allow successful reprogramming and then adopting these factors into current factor-based reprogramming methodologies.
Footnotes
Acknowledgments
The author thanks Don Wolf and Shoukhrat Mitalipov for their assistance in generating the rhesus monkey SCNT-ESC transcriptional data. I am also indebted to Jessica Byrne for creating the artwork in ![]() and for her editorial review of this manuscript. This work was supported by funding from the Broad Stem Cell Research Center at UCLA (#441458-BJ-42999).
and for her editorial review of this manuscript. This work was supported by funding from the Broad Stem Cell Research Center at UCLA (#441458-BJ-42999).
Author Contribution Summary
James Byrne performed all aspects of this transcriptional analysis study (conception and design, data analysis, manuscript writing, and final approval of manuscript).
Author Disclosure Statement
The author has no potential conflicts of interest to disclose.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.