
Abstract
Abstract
Pluripotential stem cells from livestock offer an exciting prospect for the biotechnology industry. Applying strategies established for the derivation of murine induced pluripotential stem cells (iPSCs), we have isolated ovine iPSCs that can give rise to cells characteristic of all three germ cell layers both in vitro from embryoid bodies and in teratomas in vivo. Furthermore, although at a low level, these ovine iPS cells can contribute to live-born chimeric lambs. Colonies derived from ovine embryonic fibroblasts transfected with murine cMyc, Klf4, Oct4, and Sox2 displayed smooth domes with sharp edges when grown in human embryonic stem cell (ESC) medium but not in mouse ESC medium. These ovine iPSCs were alkaline phosphatase positive, expressed Nanog, and had a normal karyotype. These cells represent an important step in the understanding of mechanistic nature of pluripotency in ungulates.
Introduction
The production of iPS cells from livestock species is attractive within an animal biotechnology setting (Montserrat et al., 2011; Roberts et al., 2009). Such cells could be of use in the generation of transgenic livestock and in modeling of stem cells treatment strategies. In this report we describe the derivation of ovine iPSCs that can give rise to cells characteristic of all three germ cell layers both in vitro and in vivo. Furthermore, although at a low level of contribution, these ovine iPSCs can contribute to live-born chimeric lambs.
Materials and Methods
Cell culture
Ovine embryonic fibroblasts (OEFs) and mouse embryonic fibroblasts (MEFs) were isolated from an early gestation fetus (13.5-day fetus for MEFs, 30-day fetus for ORFs) and cultured in 0.1% gelatine-coated plates (gelatine from porcine skin; Sigma, St. Louis, MO) in medium composed of DMEM (Sigma), 10% fetal calf serum (FCS) (PAA). SNL cells [SNL 76/7 cells, European Collection of Cell Cultures (ECACC)] were expanded on 0.1% gelatine-coated plates in DMEM, 10% FCS, 2 mM L-glutamine (Invitrogen, Carlsbad, CA), and mitotically inactivated by irradiation at 10,000 rad. Ovine iPSCs were derived on a γ-irradiated SNL feeder layer employing two different media: the first we termed human ESC medium, was composed of knock-out DMEM (Invitrogen), 20% knock-out serum replacement (Invitrogen), 2 mM L-glutamine, 0.1 mM nonessential amino acids (Gibco, Grand Island, NY), 0.1 mM 2-mercaptoethanol (Gibco), 8 ng/mL human basic fibroblast growth factor (bFGF) (Peprotech, Rocky Hill, NJ); the other we termed mouse ESC medium, was composed of DMEM, 15% FCS, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 0.1 mM 2-mercaptoethanol, 1000 U/mL mouse LIF (ESG1107, Millipore, Bedford, MA). Mouse ESCs were maintained on an SNL feeder layer in mouse ESC medium. Human ESCs were cultured on Matrigel-coated plates (Matrigel, Becton Dickinson, Lincoln Park, NJ) in filtered conditioned medium supplemented with 4 ng/mL human bFGF. To prepare conditioned medium MEFs were grown in a 150-cm2 flask in DMEM and 10% FCS, and MEF medium was then replaced with 50 mL of serum-free human ESC medium (80% knock-out DMEM, 20% knock-out serum replacement, 0.1 mM nonessential amino acids, 2 mM L-glutamine, 0.1 mM 2-mercaptoethanol, and 4 ng/mL bFGF); conditioned medium was collected from the flask the following day and stored at −20°C. Human embryo kidney cell lines (HEK 293T/17, ATCC, Rockville, MD) were grown in DMEM, 10% FCS, 2 mM L-glutamine, 0.1 mM nonessential amino acids. Cells were grown at 37°C, 5% CO2. Medium was renewed daily for ovine iPSCs, mouse ESCs, and human ESCs, and every 2–3 days for MEFs, OEFs, SNL cells, and HEK cells. Ovine iPSCs, mouse ESCs and human ESCs were passaged every 2 to 4 days as follows: the cells were fed 2–3 h before the procedure and then trypsinized employing trypsin–EDTA (Gibco) at a ratio between 1:3 and 1:5.
Virus packaging and transduction
The reprogramming plasmids were the same as used in the first iPSC report (Takahashi and Yamanaka, 2006) and were purchased from Addgene (pMXs-c-Myc Addgene plasmid 13375, pMXs-Klf4 Addgene plasmid 13370, pMXs-Oct3/4 Addgene plasmid 13366, pMXs-Sox2 Addgene plasmid 13367). The Green Fluorescent Protein (GFP) expressing plasmid employed as a control during the reprogramming (pCLXSN-GFP) was built cloning the GFP open reading frame cut with EcoRI and XhoI from the pRSET/EmGFP plasmid (Invitrogen) into the same restriction sites of the pCLXSN expression vector (Imgenex, San Diego, CA). For the reprogramming, the MoML viral vectors carrying cMyc, Klf4, Oct4, and Sox2 were individually packaged and transduced as follows: 3×106 HEK cells were plated in a 10-cm dish in 10-mL medium; when at 70% confluent (normally the day after) the cells were transfected with 8 μg of packaging plasmid (pCL-10A1 for ovine cells, pCL-Eco for mouse cells, both from Imgenex) and 8 μg of viral vector (pMXs-c-Myc, pMXs-Klf4, pMXs-Oct3/4, or pMXs-Sox2) using FuGENE HD (Roche, Indianapolis, IN) according to the manufacturer's specifications. After 24 h medium was renewed. A day later the supernatant (6 mL) was collected and filtered; 1 mL of each virus, supplemented with 4 μg/mL Polybrene (Sigma) was added to each well of a six-well plate containing OEFs seeded the day before at 1.3×105 cells/well. Six microlitres of medium were added again to the packaging plates and 24 h later the transduction was repeated again. The same protocol was employed in MEFs as a control for the reprogramming strategy and in OEFs with the GFP viral vector instead of the four factors as a control for the transduction efficiency in ovine cells. PGK-GFP (pRRL-PGK-eGFP-WPRE, Genethon, France) virus was transduced in ovine iPS cells when cells were subconfluent: the medium was replaced and after 3 h the cells were trypsinized, the feeders were removed, and the cells were split 1:3 and plated in a well of a 12-well plate on feeders with 200 μL of medium supplemented with 25 μL of virus. After 3 h 2.5 mL of medium were added and the day after medium was renewed.
PCR and RT-PCR analyses
All PCRs were performed employing Taq DNA polymerase from Roche. PCR was performed on genomic DNA (gDNA) with a primer set specific for enJSRVs; using 95°C for 10 min followed by 35 cycles composed of a denaturation step at 95°C for 30 sec, an annealing step at 59°C for 30 sec, and an elongation step at 72°C for 30 sec. Myc, Klf4, Oct4, and Sox2 primer pairs specific for the viral cassettes were employed on gDNA and on cDNA using PCR conditions: 95°C for 5 min followed by 35 cycles composed of a denaturation step at 94°C for 1 min, an annealing step at 65°C for 1 min and an elongation step at 72°C for 1 min. For genotyping, primers sets specific for the viral Oct4, cMyc, and Klf4 were employed using these PCR conditions: 95°C for 5 min followed by 45 cycles composed of a denaturation step at 94°C for 1 min, an annealing step at 65°C for 1 min and an elongation step at 72°C for 30 sec. Ovine HPRT primers were used as a control to verify the presence and the quality of cDNA and gDNA; the PCR conditions applied were: 95°C for 5 min followed by 30 cycles composed of a denaturation step at 94°C for 1 min, an annealing step at 55°C for 1 min, and an elongation step at 72°C for 30 sec. Reverse transcription was carried out on DNase-digested RNA with oligo(dT)20 using Superscript III First-Strand Synthesis System (Invitrogen) according to the manufacturer's specifications.
Alkaline phosphatase (AP) staining and immunocytochemistry
For AP staining and for immunocytochemistry, cells were prepared removing the medium, washing them twice in phosphate-buffered saline (PBS), fixing them in 4% paraformaldehyde (PFA, Sigma) for 20 min at room temperature, and washing again twice in PBS. AP staining was performed with Leukocyte Alkaline Phosphatase Kit (Sigma) as follows: 0.2 mL of Sodium Nitrite Solution were added to 0.2 mL of FRV-Alkaline Solution, mixed gently, and allowed to stand for 2 min; the solution was then combined with 9 mL of ddH2O and 0.2 mL of Naphthol AS-BI Alkaline Solution were also added and mixed thoroughly. A total of 0.35 mL of final solution were then added to each well of a 24-well plate and let to stand overnight in the dark and rinsed after 24 h with PBS. For immunocytochemistry against intracellular markers cells were permeabilized for 20 min at room temperature. For both intracellular and surface markers, cells were blocked from 1 to 2 h at room temperature. Primary antibodies were diluted in the blocking solution and applied to the cells overnight at 4°C. Plates were then washed four times for 5 min in washing buffer and blocked again for 15 min at room temperature. The appropriate FITC conjugated secondary antibody was diluted in washing solution (in blocking buffer for Nanog) and added to the cells for 1 to 2 h at room temperature in the dark. Cells were later washed four times for 5 min in PBST and the nuclei were stained with 1 μg/mL of DAPI (Sigma).
In vitro differentiation
When around 70% confluent, cells were trypsinized, but not at single-cell suspension; small clusters of cells were obtained and plated in gelatine-coated plates for 20 min to separate them from the feeder layer. Ovine iPSCs were later collected and seeded in low attachment culture dishes in human ESC medium. The day after, the generated embryoid bodies (EBs) were washed and medium replaced with differentiation medium (knock-out DMEM, 10% FCS, 2 mM L-glutamine, 0.1 mM nonessential amino acids, and 100 μM 2-mercaptoethanol). EBs were allowed to grow and were washed every other day. After 4 to 7 days of culture the EBs were transferred onto adherent, gelatine-coated plates and cultured in differentiation medium until well-developed outgrows appeared. Cells were then washed twice in PBS, fixed for 20 min with 4% PFA, and stained for markers of differentiation.
Teratoma formation and immunohistochemistry
Ovine iPSCs resuspended in 100 μL PBS were intramuscularly injected in the right leg of severe combined immunodeficiency (SCID) mice (106 cells/mouse). After 3 to 5 weeks, mice were sacrificed. Tumors were collected and either enclosed in OCT embedding medium (Tissue-Tek, Dublin, OH) and fixed in liquid nitrogen for 2 h or fixed in a formal saline solution (ddH2O, 4% formaldehyde, 0.9% NaCl) and paraffin embedded. Sections were stained against markers of differentiation and histologically analyzed with the same antibodies employed for the immunocytochemistry: section were cut, fixed in acetone for 2 min at room temperature and rinsed in 0.1 M PBS for 2 min. They were postfixed in fresh Periodate Lysine Paraformaldehyde solution for 8 min at room temperature, rinsed in PBS for 2 min, rinsed in distilled water for 2 min, and transferred to Sequenza coverplates using filtered TBST (Tris-buffered saline pH 7.5, Tween 20). The sections were later washed twice in TBST, incubated with diluted primary antibody for 1 h at 25°C, rinsed three times in TBST, and blocked with 100 μL Dako Real blocker for 20 min at room temperature. The slides were washed again three times in TBST and 100 μL of Secondary link polymer complex (Dako Envision kit) were added for 30 min at 25°C. The sections were washed five times in TBST and allowed to drain completely. They were then rinsed in distilled water and allowed to drain completely. A total of 100 μL of DAB chromogen was added twice for 5 min at 25°C and the sections were washed once in TBST. Counterstaining was carried out with hematoxylin using the Gemini autostainer.
Metaphase spreads
Colcemid (Invitrogen) at a final concentration of 0.1 μg/mL was added to cells and left for 2 h at 37°C. Cells were later rinsed with PBS and dissociated with trypsin. PBS was added to dilute the trypsin and cells were centrifuged at 250×g for 8 min. Supernatant was removed and the pellet was resuspended in 10 mL 75 mM KCl (Sigma) and incubated for 15 min at 37°C. A total of 1 mL of ice cold 3:1 methanol/acetic acid was added and cells were centrifuged at 200×g for 5min. Supernatant was removed and the pellet was resuspended in 5 mL of ice cold 3:1 methanol/acetic and centrifuged at 200×g for 5 min; this step was repeated three times. Cells were later resuspended in 1 mL 3:1 methanol/acetic acid and spread on a glass microscope slide refrigerated at 4°C in 70% ethanol. Slides were left at 70–80°C overnight and stained with a 10% Giemsa solution (Sigma).
Population doubling time
Ovine iPSCs were seeded at low density (3.55×104 cells per well of a 48-well plate) and counted in triplicates every 12 h for 96 h. Using MiniTab a linear regression was performed on the exponential phase of the growth.
Ovary collection and oocyte manipulation
Ovine ovaries were collected at the abattoir, maintained at 30°C during transport to the laboratory, and washed in warm PBS. Follicular fluid was collected using 18-gauge needle and 10-mL syringe and placed into 15-mL centrifuge tubes containing fresh warm wash medium (1×Medium 199, 4.76 mM NaHCO3, 12.5 mM HEPES, 112.5 U/mL Heparin, 10.875 μL/mL estrus sheep serum, H2O, pH=7.4; all reagents from Sigma). Good-quality oocytes surrounded by a compact cumulus mass were selected and washed again three times in the wash medium and once in fresh maturation medium (1× Medium 199, 6.25 mM NaHCO3, 2 mM L-glutamine, 5 ng/mL ovine FSH, 5 ng/mL ovine LH, 0.5 ng/mL estradiol, 250 μL/mL estrus sheep serum, H2O, pH=7.6; all reagents from Sigma). Oocytes were placed in 800 μL of maturation medium in a four-well Nunc tissue culture plate (40–50 oocytes/well) and incubated for approximately 22 h at 5% CO2, 38.5°C. After this time, oocytes, which have been matured to MII stage of development, are prepared for fertilization removing cumulus cells by pipetting in wash medium. Oocytes surrounded by coronal cells were washed twice in fertilization medium composed of synthetic ovidictal fluid (SOF) (Walker et al., 1996) 2% estrus sheep serum, pH=7.8. The oocytes were then fertilized with the upper fraction of ram semen pellets stored in liquid nitrogen and previously thawed and activated using a swim up technique. Cells were then incubated at 5% CO2, 38.5°C and after 22–24 h were washed twice in warm SOFaaBSA medium [SOF medium, 1× BME amino acids solution, 1× MEM nonessential amino acids solution, 2 mM L-glutamine, 4 mg/mL bovine serum albumin (BSA), pH=7.4; all reagents from Sigma], removing the sperm and any remaining cumulus cells by gently pipetting. Fertilized oocytes were then placed into preequilibrated four-well plates in SOFaaBSA medium and overlaid with mineral oil and culture in a 5:5:90 incubator at 38.5°C. After 24 h cells were checked to assess development and remove any that have not cleaved.
Injection of ovine iPSCs in early-stage embryos
Ovine iPSCs ranging from passage 8 to passage 23 were microinjected into zygotes, eight-cell embryos, day 6 or day 7 blastocysts as follows: starting from one subconfluent well of a six-well plate (50–70% confluent), 2–3 h before harvesting the medium was changed, ovine iPSCs were trypsinized to obtain a single cell suspension and plated in a gelatine-coated T25 cm2 flask. In order to separate the cells from the feeder layer, the flask was placed for 20 min in the incubator at 37°C, 5% CO2. Immediately prior to microinjection, cells were collected, centrifuged, resuspended in 500 μL of medium, and stored at room temperature until microinjection was complete.
Transfer of injected blastocysts in the recipient
Following microinjection, zygotes and eight-cell embryos were culture until blastocyst stage in 850-μL drops of SOFaaBSA under mineral oil at 38.5°C in an atmosphere of 5% CO2:5% O2:90% N2. If these embryos developed into blastocysts they were then transferred in recipient ewes. Blastocysts that were microinjected were transferred into recipient ewes on the same day or on the day after the microinjection. Recipient ewes were selected from groups of cycling adults in which estrus was synchronized using intravaginal progestagen sponges (Veramix, Upjohn, Kalamazoo, MI). Estrus was detected by introduction of vasectomized males. Animals that had exhibited estrus 6 or 7 days previous to transfer were selected as recipients for blastocysts. Following a general anesthetic a midline laparotomy was performed to expose the uterine tract. A small puncture was made in the uterine wall using a blunt 16-gauge needle and a 20-mL Drummond-positive pipette fitted with sterile “fire-polished” capillaries were used for the transfer. The animals were monitored throughout pregnancy by transcutaneous ultrasound examination.
Results
Induction of pluripotency in ovine cells
OEFs (Fig. 1A) were transduced with four MoML viral vectors, each carrying a different reprogramming factor (cMyc, Klf4, Oct4, and Sox2). We employed the murine versions of the genes, which are widely reported to be able to reprogram also cells from other animals, despite cross-species differences (Esteban et al., 2009; Mali et al., 2008). Two days after transduction, cells were passaged onto γ-irradiated SNLs. At day 4, two different culture conditions were applied: mouse and human ESC medium. The former contains FCS and mouse LIF, which are replaced with serum replacement and human bFGF in the latter. At day 8, small round aggregates of cells started appearing in both media and over the following few days some of the cell clumps progressed into clusters of cells, revealing differences between the two conditions. In mouse ESC medium the colonies looked granulated, without a precise segregation from the layer of parental fibroblasts, suggesting that the reprogramming was not complete. In contrast, in human ESC medium the colonies showed a more ESC-like morphology, with sharp edges and smooth domes (Fig. 1B and C).
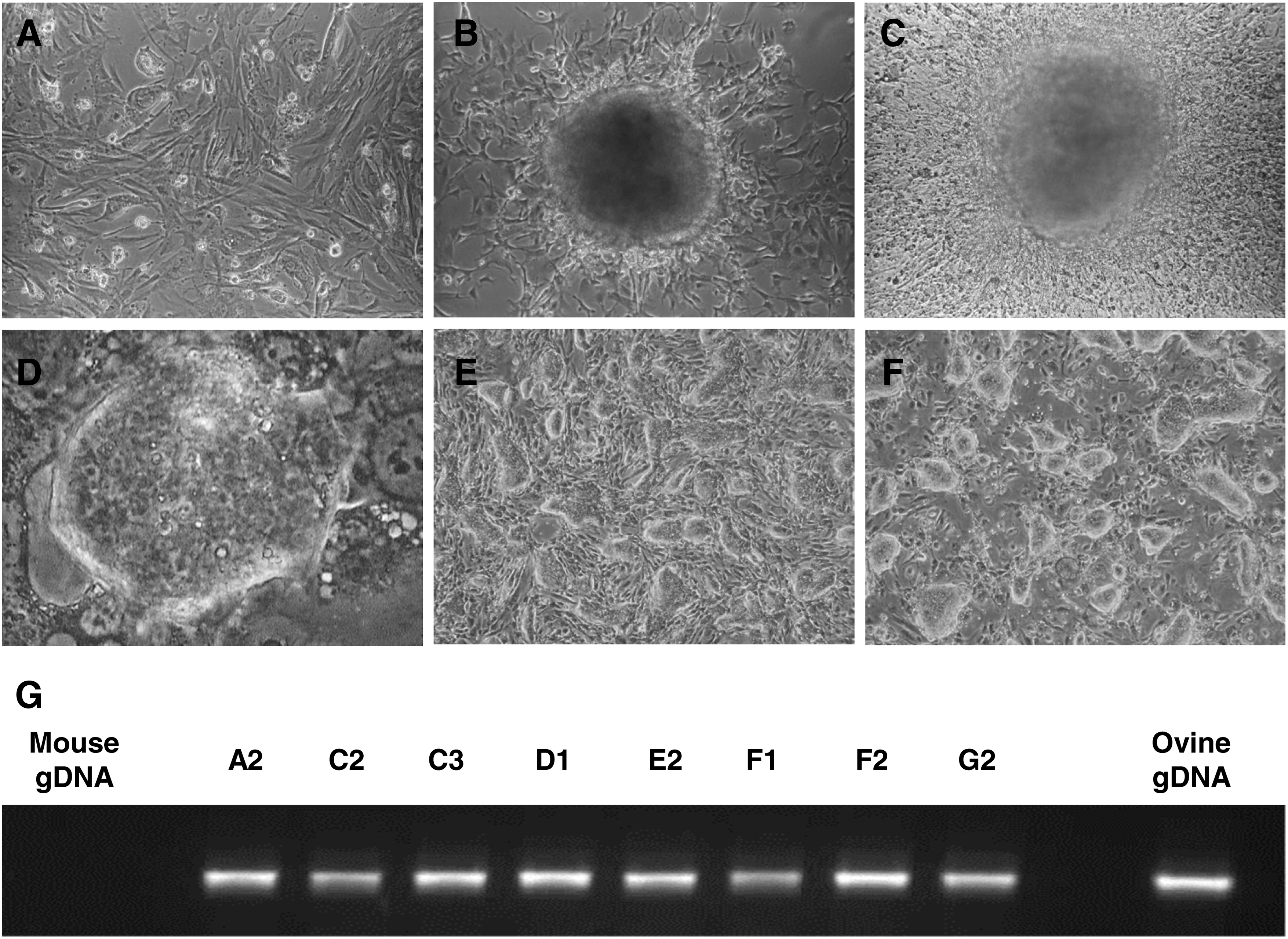
Ovine iPSCs derived from embryonic fibroblasts.
A total of 13 colonies were picked from the human ESC condition group and expanded. During this derivation phase, clones were grown on SNL feeder cells in human ESC medium and passaged with trypsin. Among the picked clones, some stopped growing, some started differentiating, while others flattened and acquired a very high proliferation rate. Only a few clones (3/13: D1, F2, and G2) maintained the typical traits of mouse ESCs, displaying compact colonies with defined borders and an even surface. These colonies were characterized by small, round cells with a high nuclear/cytoplasmic ratio (Fig. 1D and E) and could be maintained for many passages without loss of phenotype (Fig. 1F); we have maintained G2 cells for over 70 days (equivalent to 25 passages). Compared to the parental OEFs, these colonies displayed greater growth rates requiring to be passaged every 2–4 days at a ratio between 1:3 and 1:5.
Because we have utilized mouse feeders in our derivation protocol, we performed a sheep-specific PCR assay to confirm the identity of the iPSCs. Primers were selected specific for the endogenous Jaagsiekte sheep retrovirus (enJSRV), which is a provirus naturally integrated in the Ovis aries genome (Dunlap et al., 2006). As shown in Figure 1G, this PCR confirmed that all the clones analyzed were indeed of sheep origin.
To assess whether the transduced four genes are transcriptionally silenced during the derivation process, we carried out RT-PCR using primers specific to the integrated constructs on RNA isolated from passage 7 cells. In order to discriminate between transgenic (mouse) and endogenous (ovine) transcripts, a common forward primer was designed in the retroviral vector, while single reverse primers were designed to be specific to each of the four mouse genes. Most clones still retained the transgene specific RNA, while one (D1) retained a residual low level of Oct4 transcripts and another (G2) displayed very low Oct4 transcripts (Fig. 2A); this analysis, however, does not differentiate between all cells displaying very low Oct4 transcription or a heterogeneous population where only a few cells retain Oct4 expression. PCR performed with the same primers on genomic DNA confirmed that all four transgenes were integrated in these two clones (Fig. 2B).
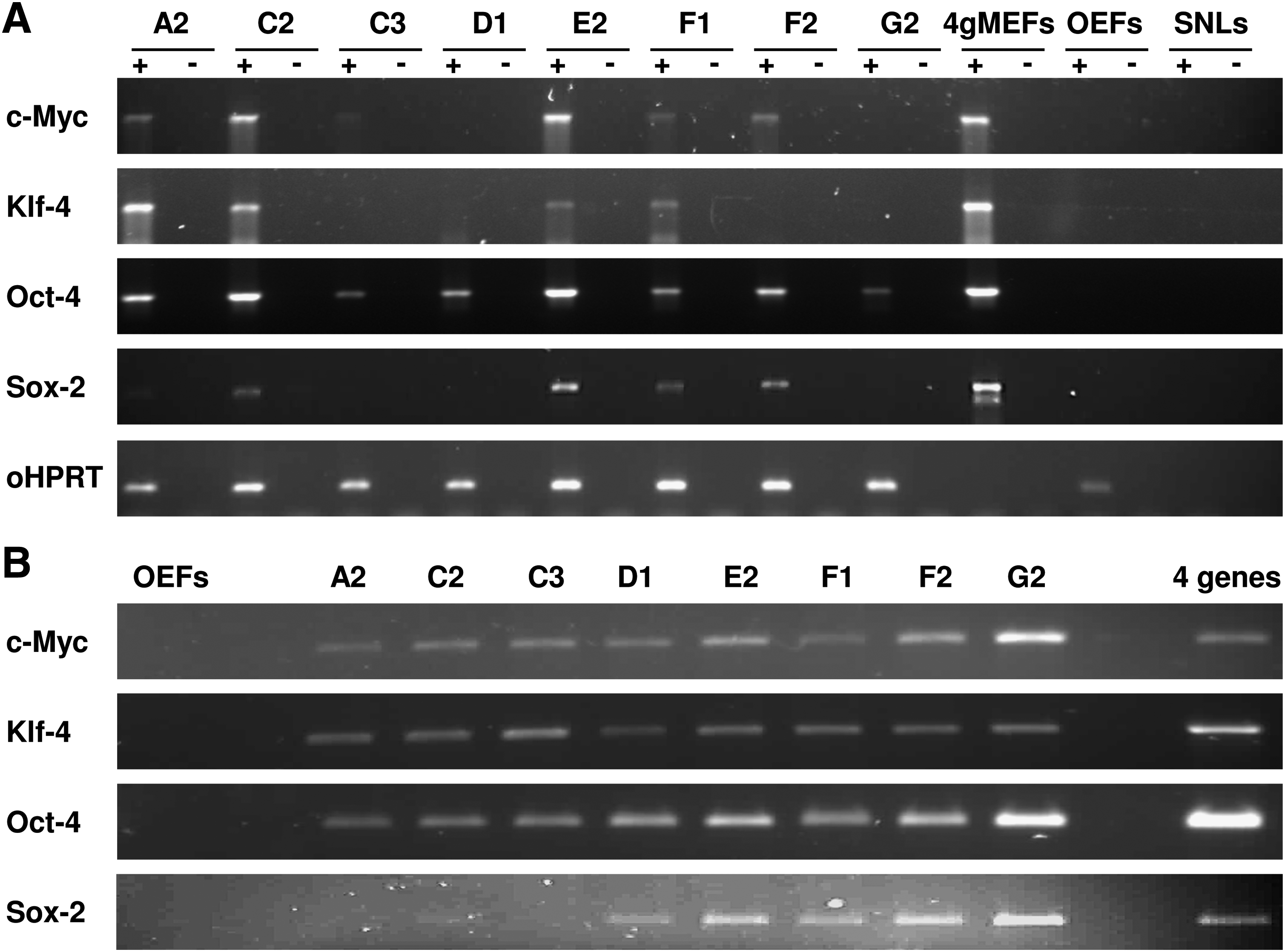
Integration and silencing of the transgenes in the reprogrammed cells.
Characterization of ovine iPSCs
Induction of pluripotency correlates with activity of specific biomarkers. First, we stained the ovine iPSC clones for alkaline phosphatase, one of the first markers to appear during the reprogramming process (Brambrink et al., 2008). Most of the clones were alkaline phosphatase negative, whereas D1 displayed weak staining and G2 gave a strong signal (Fig. 3A). Although alkaline phosphatase activity marks the initial phases of reprogramming, Nanog expression is associated with the pluripotent state in mouse iPSCs (Brambrink et al., 2008). G2 cells stained positive for Nanog and the signal was restricted to the nucleus (Fig. 3B). This initial analysis supports the premise that G2 cells are reprogrammed to a self-renewing state; however, verification of this conclusion will require a more thorough characterization of a range of pluripotency markers. All the tested clones, passage 9, were on the whole negative for the pluripotency markers SSEA1 and SSEA4, with only a small number of cells in only few clones displaying staining for these markers (Fig. 3C).
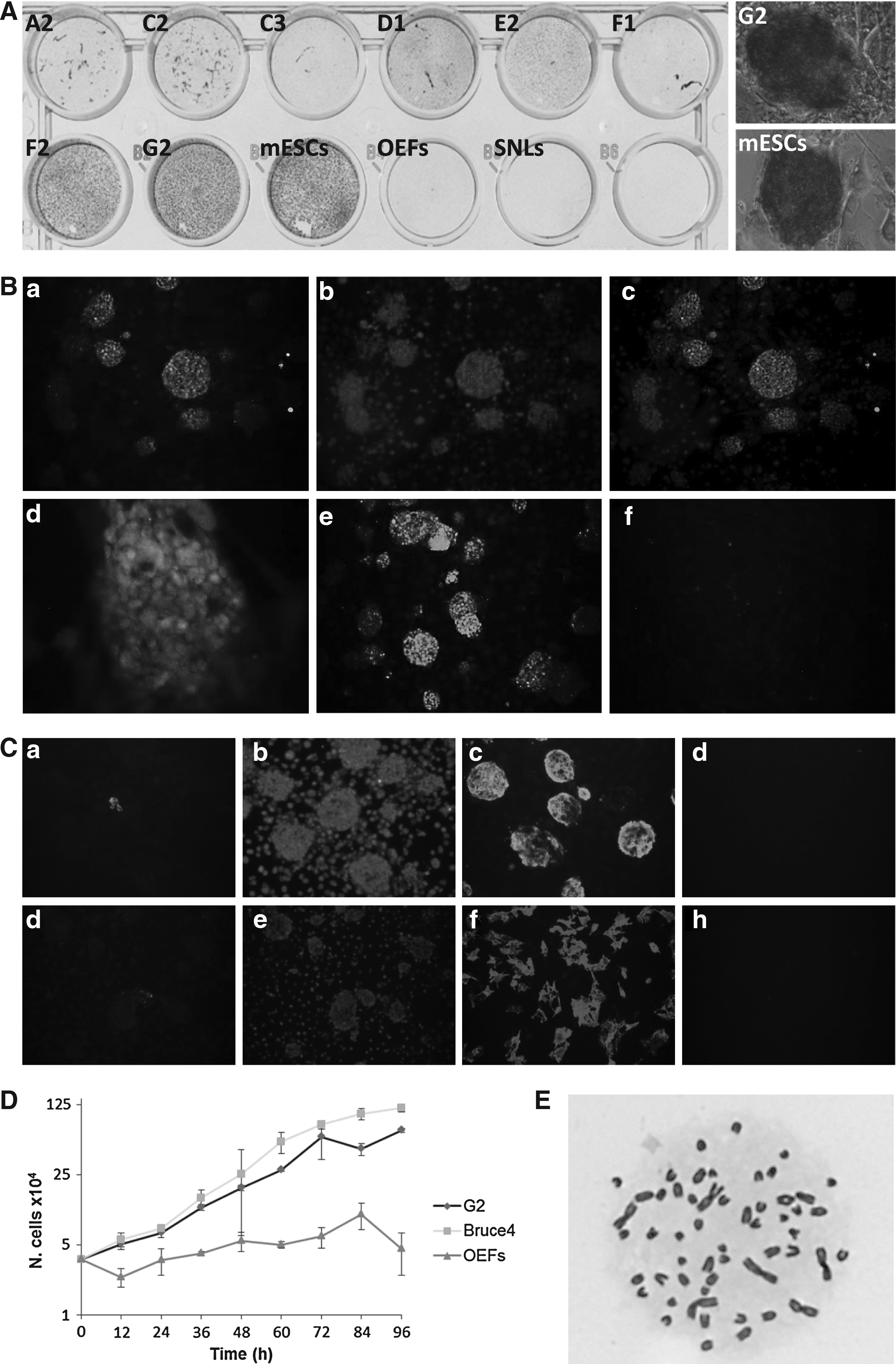
Ovine iPSCs show characteristics similar to ESCs.
Another significant change occurring during reprogramming is the increase in proliferation, with iPSCs acquiring a population doubling time similar to that of ESCs (Berrill et al., 2004). This is indeed what we observed for our ovine iPSCs: a population doubling time of 13.8 and 17.5 h for murine ESCs and ovine G2 iPSCs, respectively, in comparison to 63 h for the parental OEFs as determined from growth curves (Fig. 3D). In addition, analyses of the G2 clone indicated the majority of cells had normal chromosome counts, with 83% of cells having 54 chromosomes (Fig. 3E); 4% with 53, 3% with 52, 3% with 51, 2% 50, 1% with 49, 2% with 48, 1% with 46, and 1% with 45 chromosomes.
Ovine iPSCs can differentiate into cells of all three germ layers
Given the desired characteristics of the G2 cells, we evaluated their potential further. G2 cells were trypsinized in order to produce small clumps of cells, separated from the feeder layer and grown in a low attachment plate in differentiation medium. After 5 days, the G2 cells formed the smooth, spherical, floating embryoid bodies (Fig. 4A) typical of ESC differentiation (Desbaillets et al., 2000; Doetschman et al., 1985), while the other clones attached to the bottom of the plate or generated floating aggregates with irregular shape (data not shown). Spontaneous in vitro differentiation was evident from these embryoid bodies after plating them onto gelatine-coated plates (Fig. 4A and B): the G2 cells stained positive for vimentin (mesoderm), βIII tubulin (ectoderm), α fetoprotein, and cytokeratin-18 (endoderm). Undifferentiated G2 cells were negative for these markers (Fig. 4C).
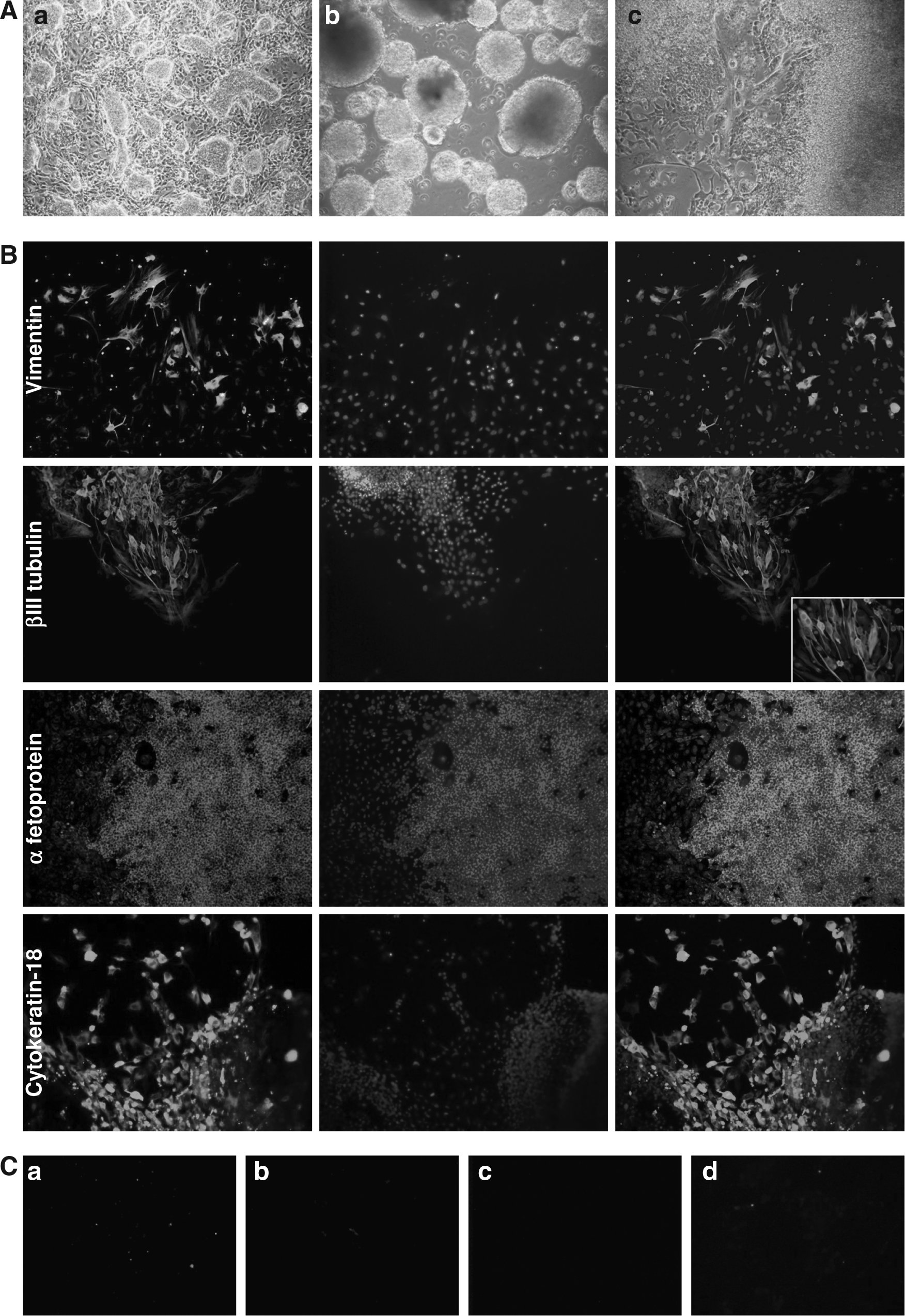
In vitro differentiation of ovine iPSCs
To assess the pluripotency of the generated ovine iPSCs in vivo, G2 cells were intramuscularly injected into SCID mice. Due to the lack of antibodies specific for ovine tissues, in order to prove that the tumors were derived from sheep, the experiment was performed inoculating GFP transduced cells (Fig. 5A). After injection, all mice developed a tumor and were sacrificed between 3 and 5 weeks later because of the size of the tumor (timing comparable with that of mouse ES cells). Brightly fluorescing green areas were noticeable in the bulk of all tumors, confirming they were derived from the injected ovine iPSCs. The fluorescence, however, was not uniform: it was also possible to find regions where GFP absent (Fig. 5B). These tumors stained positive to vimentin, βIII tubulin, α fetoprotein, and cytokeratin-18 (Fig. 5C–F); importantly, there was colocalization of GFP fluorescence and cell-type staining patterns (Fig. 5B and C).
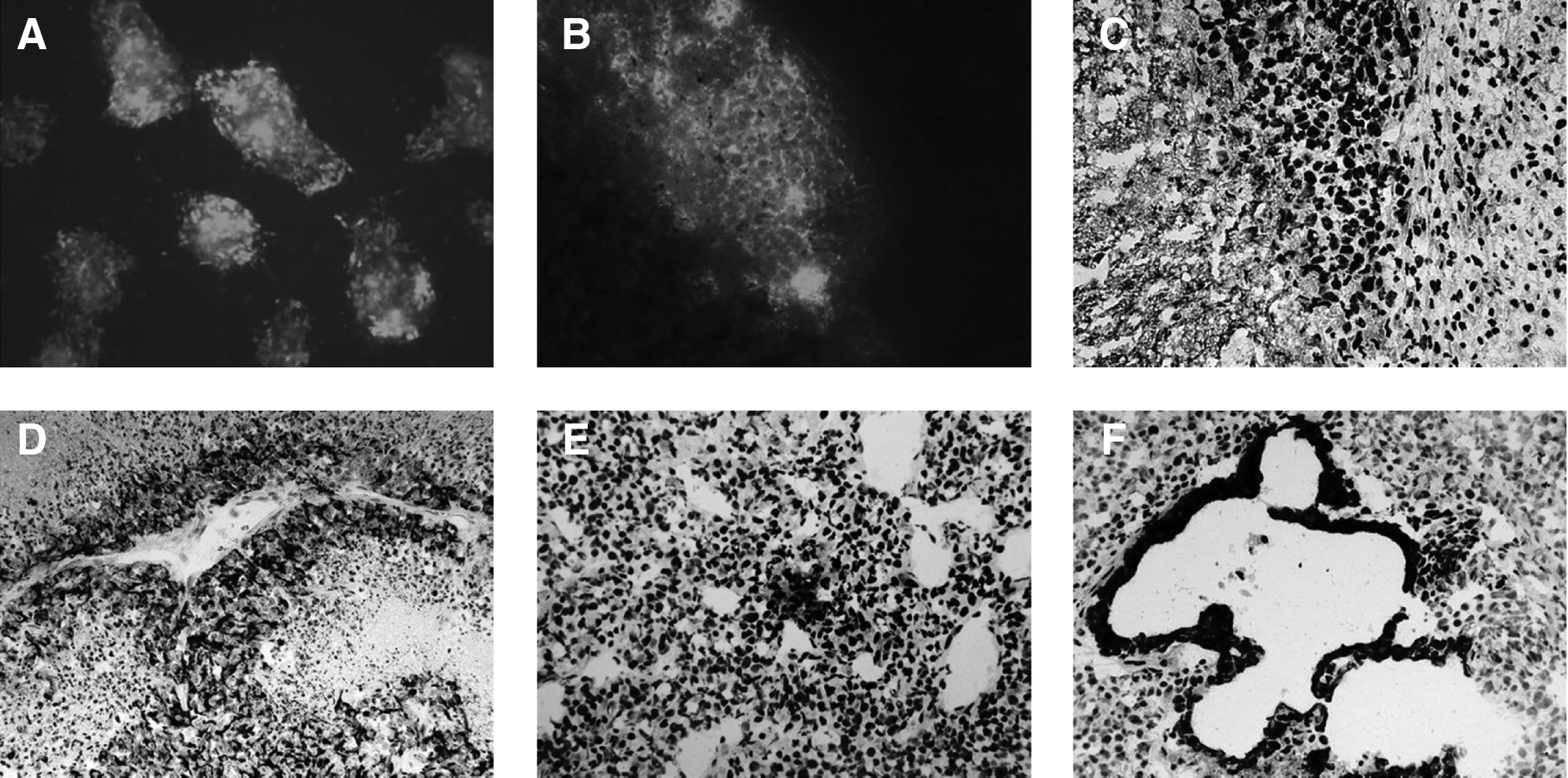
Teratoma formation and analyses. Injections were performed with ovine iPSCs at passage 9 and passage 18. The figure shows the GFP fluorescent ovine iPSCs employed for the in vivo differentiation assay
Finally we evaluated the ability of G2 cells to contribute to the production of chimeric animals. G2 cells were injected into zygotes, eight-cell stage embryos, or blastocysts (Table 1) and transferred into synchronized pseudopregnant recipient ewes when at blastocyst stage. Three recipient ewes were sacrificed at 3 weeks postfertilization and PCR performed on genomic DNA. In the six embryos recovered a reproducible but faint PCR detection of Oct4 fragment was observed (Fig. 6A). This suggested that the embryos contained iPSC derived cells, but that these constituted only a small proportion. The remaining ewes were allowed to carry the pregnancy to term with 17 live lambs and one stillborn delivered. For some of the ewes gestation term was a few days longer than expected and three lambs were very big, with a weight ranging between 7.5 and 10.2 kg, whereas one was very small, weighting only 2 kg. The lambs were sacrificed (from 5 to 16 weeks of age) and multiple samples were collected from a range of tissues and subjected to PCR specific for the transgene encoding Oct4 and cMyc. The Oct4 results showed a very low iPSC contribution in different tissues (about one iPSC in 103 cells or lower). This was confirmed by cMyc PCRs in few samples (Fig. 6B).

Ovine iPSCs generate chimeras. PCR on fetuses
Ewes sacrificed 3 weeks after fertilization; nine fetuses were recovered.
One mummified lamb.
Discussion
The derivation of iPSCs from species other than mouse and human is still in its infancy. Initial attempts to derive porcine iPSCs have encountered variable reprogramming (Esteban et al., 2009; Ezashi et al., 2009; West et al., 2010; Wu et al., 2009), whereas the first two reports of ovine iPSCs describe contrasting results; one indicating that ovine iPSCs can be derived (Li et al., 2011) using the original four genes described (Takahashi and Yamanaka, 2006), while the other failed to derive iPSCs (Bao et al., 2011) following this strategy. In our hands we were able to derive ovine cells displaying many characteristics of iPSCs using the original four-gene method. These cells display markers of the three lineages after differentiation from embryoid bodies in vitro and in teratomas in vivo. Furthermore, we show for the first time that ovine iPSCs can contribute, albeit at a low frequency, to live-born lambs.
The derivation of pluripotent cells in sheep has always been challenging: the isolation of ESC-like cells from ovine blastocysts has been described (Dattena et al., 2006), but these cells could not be maintained in an undifferentiated state. In addition to our data, two recent reports describe the generation of ovine iPSCs; distinct differences exist between our data and that published by Li et al. (2011) and Bao et al. (2011), specifically relating to what genes were required for derivation, whether maintenance of expression of the transduced genes was required and what the expression signature of pluripotency markers that have been describe for mouse and human iPSCs is. We discuss these aspects in turn below.
The first iPSCs were derived using only four genes; Oct4, Sox2, Klf4, and cMyc (Takahashi and Yamanaka, 2006). Subsequently iPSCs from other species have been derived using these four genes (Esteban et al., 2009; Honda et al., 2010; Li et al., 2009). Although reported by Bao and colleagues (2011) that this strategy was not sufficient for the derivation of ovine iPSCs, our data supports that of Li and colleagues (2011) in showing that ovine cells displaying iPSC characteristics can be derived using only the original four genes. Given that iPSCs have now been derived using the four-gene strategy for a number of species, it is likely that early embryonic signaling events, or at least signaling events associated with the formation of pluripotency in isolated cells, are common across a range of mammalian species. Indeed, it may be that pluripotency in all mammalian species reflects a common signaling mechanism, a concept supported by recent progress in nonmurine ESC culture (Buehr et al., 2008).
When working with retroviral vectors the observation in mouse and human cells is that over time the transduced genes are silenced through a mechanism reflected in DNA methylation (Hotta and Ellis, 2008). Subsequent strategies engineer this silencing process into constructs through the use of inducible promoters such as that based on the TET system (Wernig et al., 2008). After silencing, a fully induced pluripotent cell will continue to self-renew given the appropriate culture environment (Okada et al., 2010). In contrast, the inability to silence the transduced genes results in cells that have considerably impaired differentiation capacity (Brambrink et al., 2008), while studies in which silencing is associated with loss of self-renewal indicated only partial reprogramming (Hotta and Ellis, 2008). We observed silencing of Klf4 and Sox2. In addition Oct4 expression was significantly downregulated but not entirely silenced. Importantly, we observed silencing of cMyc; thus, the increased proliferation and morphologically less distinct teratomas are unlikely to be simply due to the transforming properties of cMyc. Thus, we observe nearly complete silencing of the induced genes while still retaining an undifferentiated phenotype and maintenance of markers of pluripotency in contrast to the previous reports on ovine iPSCs (Bao et al., 2011; Li et al., 2011), regardless of which induction strategy was employed. This difference could reflect differences in the parental cells used, fetal or young adult, and/or the genotype of sheep from which they were derived. Alternatively, each study utilized slightly differing culture conditions that could account for the observed differences. It is more likely, however, that this difference is simply a reflection of our use of retrovirus-based vectors in contrast to lentivirus vectors. Although at this stage it is not clear as to why this difference exists, it may correspond to the ability of our ovine iPSCs to contribute to live-born lambs.
Pluripotency is associated with the expression of a specific set of genes (Koestenbauer et al., 2006) and although largely similar, differences between species (mouse and human) exist, for example, SSEA1 is expressed only in mouse and SSEA4 only in human (Koestenbauer et al., 2006). Our ovine iPSCs expressed some of these markers. Importantly, they expressed the key pluripotency Nanog (Mitsui et al., 2003), but we failed to detect uniform expression of either SSEA1 or SSEA4. This differs from the other ovine iPSC reports where either SSEA1 or SSEA4 were detected. Again, it is not clear as to why such expression difference exist for ovine iPSCs. In the work of Li et al. (2011), flattened human ESC-like colonies were identified perhaps reflecting the expression of SSEA4, whereas more compact tight colonies were reported by Bao et al. (2011), where SSEA1 expression was observed. In our study we specifically selected those colonies that had a mouse ESC-like appearance. It is possible that iPSCs from different species will display a distinct but overlapping expression signature; however, given the differences in expression profile for ovine iPSCs, it is premature to draw conclusions with respect to the ovine iPSCs marker profile and to how this compares to that established for mouse and human.
Additional aspects of the reports of ovine iPSC derivation also merit comment. Our first colonies appeared 8 days after the transduction, only 2 days later than in our mouse control experiment but substantially quicker than the 14 (Li et al., 2011) and 20 (Bao et al., 2011) days reported previously. This may simply reflect differences in choice of the vector, the use of fetal versus adult fibroblasts, or subtle differences in culture conditions. Media in particular can heavily affect the dedifferentiation process: in the mouse, a higher number of iPSC colonies can be generated if the cells are reprogrammed in a medium containing KO-DMEM and SR, rather than in other media containing DMEM and FCS, DMEM, and SR or KO-DMEM and FCS (Okada et al., 2010). In accord with a previous report (Bai et al., 2008), we observed better morphology when the cells were generated in KO-DMEM and SR rather than in DMEM and FCS. In contrast, Li and colleagues (2011) observed a higher proportion of AP-positive cells in FCS; this could reflect the observed differences in colony morphology between the two studies. Furthermore AP, although being a marker for ESC cells, is not strictly specific to pluripotent cells; therefore, the higher proportion of AP-positive cells in FCS might be due to a more permissive culture condition than SR for the development of partially reprogrammed cells. This could be consistent with our observation that, transducing cells with only cMyc and growing them in the presence of either FCS or SR, a higher proportion of AP-positive colonies appeared in the FCS conditions (data not shown).
In ungulates, iPSCs able to give rise to chimeras have already been established (West et al., 2010); however, they were derived from porcine mesenchymal stem cells. We now report the first demonstration that iPSCs derived from a differentiated cell type (fibroblast) can contribute to live born animals in species other than the mouse after injection into early stage embryos. Although producing chimeric lambs, the contribution of ovine iPSCs in live-born animals was very low. Although our ovine iPSCs have a normal karyotype and show features similar to that of ESCs (such as morphology, population doubling time, and ability to differentiate in vitro and in vivo), and thus could be expected to reflect murine data, very little is known about the development of the early embryo in sheep. Specifically, there is no prior knowledge about how a pluripotent cell injected into the early ovine embryo will be incorporated. Subtle differences in growth characteristics could impact on this process. For example, even though the growth rate of our ovine iPSCs was comparable to established mouse ESCs, we observed that after trypsinization our ovine iPSCs took longer to recover than the mouse ESCs (data not shown). This delay may reduce the proliferation capability of cells injected into the embryo. The low contribution to chimeras may also be due to the culture medium in which our ovine iPSCs are grown: human ESC conditions are known to promote the epiblast stem cell (EpiSC) state both in mouse and in human. EpiSCs express the key pluripotency factors and can differentiate into numerous cell types in vitro; however, they are not able to contribute to live animals after injection into blastocysts (Tesar et al., 2007). In light of this consideration, the low contribution to the chimaeras is to be expected. Alternatively, and reflecting the limited morphology observed in teratoma studies, our iPSCs may not be fully reprogrammed. Indeed, even though displaying these marker profiles, the ovine iPSC-derived teratomas were morphologically not as distinctive as that usually observed for mouse teratomas. This might reflect that the cells are not fully reprogrammed or simply what happens when sheep cells are injected into nude mice (see Bao et al., 2011; Li et al., 2011l). Finally, the ability of ESCs (Beddington and Robertson, 1989), previously reported porcine iPSCs (West et al., 2010) and even fibroblasts (Piliszelk et al., 2007) have been reported to contribute to chimeric embryos. In conclusion, the contribution of our ovine iPSCs to live-born lambs merely represents an initial step in the process of deriving ovine iPSCs.
The generation of ovine iPSCs is an important step in the understanding of the mechanism underlying pluripotency and differentiation in ungulates. To date, attempts to derive ovine ESCs have not been successful. It is possible that protocols for derivation of ovine iPSCs could provide a platform for the screening, and the optimization of the correct culture conditions for the derivation of pluripotent cells form the sheep embryo leading to the isolation of authentic ovine ESCs. Finally, either such ovine ESCs, or ovine iPSCs derived without the use integrating vectors, may provide a valuable resource for transgenesis in this agriculturally important species.
Footnotes
Acknowledgments
Thanks to Gillian Parham for the pCLXSN-GFP plasmid, Prof. Massimo Palmarini's group for the indication of the enJSRVs primer set specific for the ovine species, Dr. Zen Lu for the help with primer design and sequence analyses, and Dr. Robert Fleming for the assistance in microscopy. We are grateful to Drs. Neil Macintyre and Audrey Graham for the processing and immunohistochemistry of teratomas, to the Large Animal Unit of the Roslin Institute for the work involved in the generation of chimeric animals, to Shirley Tong for the karyotyping, and to Claire Neil for the help with DNA extraction from ovine tissues. Thanks also to Dr. Stephen Meek, Dr. Amandine Breton, Linda Sutherland, Catalina Diaz, and Prof. Gaetano Donofrio for advice on the experimental design and comments on the work. This work was supported by an Early Stage Researcher (ERS) post within the EC-CLONET Marie Curie Initial Training Network (ITN) and through the Biotechnology and Biological Sciences Research Council (BBSRC).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.