
Abstract
Abstract
We have established a serum- and feeder-free culture system for the efficient differentiation of multifunctional hepatocytes from human embryonic stem (ES) cells and three entirely different induced pluripotent stem (iPS) cells (including vector/transgene-free iPS cells generated using Sendai virus vector) without cell sorting and gene manipulation. The differentiation-inducing protocol consisted of a first stage; endoderm induction, second stage; hepatic initiation, and third stage; hepatic maturation. At the end of differentiation culture, hepatocytes induced from human pluripotent stem cells expressed hepatocyte-specific proteins, such as α-fetoprotein, albumin, α1 antitrypsin and cytochrome P450 (CYP3A4), at similar or higher levels compared with three control human hepatocyte or hepatic cell lines. These human iPS/ES cell-derived hepatocytes also showed mature hepatocyte functions: indocyanine green dye uptake (∼30%), storage of glycogen (>80%) and metabolic activity of CYP3A4. Furthermore, they produced a highly sensitive hepatotoxicity assay system for D-galactosamine as determined by the extracellular release of hepatocyte-specific enzymes. Hepatoprotective prostaglandin E1 attenuated this toxicity. Interestingly, bile duct-specific enzymes were also detected after drug treatment, suggesting the presence of bile-duct epithelial cells (cholangiocytes) in our culture system. Electron microscopic studies confirmed the existence of cholangiocytes, and an immunostaining study proved the presence of bipotential hepatoblasts with high potential for proliferation. Differentiated cells were transferrable onto new dishes, on which small-sized proliferating cells with hepatocyte markers emerged and expanded. Thus, our differentiation culture system provides mature functional hepatocytes, cholangiocytes, and their progenitors with proliferative potential from a wide variety of human pluripotent stem cells.
Introduction
Induced pluripotent stem (iPS) cells were generated directly from somatic cells as a result of the introduction of four reprogramming factors, Oct4, Sox2, Klf4, and c-Myc (Park et al., 2008; Takahashi and Yamanaka, 2006; Takahashi et al., 2007; Yu et al., 2007), and shared many characteristics with ES cells, including multilineage differentiation potential, and intensive proliferation in vitro. In addition, the establishment of human iPS cells is ethically acceptable and does not require human oocytes. Thus, we may be able to obtain patient-specific functional cells for research into diseases, apply these cells to the regenerative medicine for therapeutic use, and use these cells for in vitro testing to satisfy industrial requirements, including drug discovery. However, as is the cases with human ES cells, it is not easy to regulate the differentiation of human iPS cells toward endoderm cells such as hepatocytes (Inamura et al., 2011; Liu et al., 2010; Si-Tayeb et al., 2010; Song et al., 2009; Sullivan et al., 2010). In addition, unlike human ES cells, most of human iPS cells have been generated via retrovirus/lentivirus vector systems, resulting in genomic integration of viral components into iPS cells (Park et al., 2008; Takahashi and Yamanaka, 2006; Takahashi et al., 2007; Yu et al., 2007).
Here we report the establishment of a serum- and feeder-free method for hepatocyte differentiation of human iPS/ES cells (including virus-free iPS cells established using Sendai virus vector) (Fusaki et al., 2009), providing excellent tools for the evaluation of drug metabolism and hepatotoxicity. We succeeded in producing cholangiocytes and bipotential hepatoblasts and developed minimally invasive subculture methods of proliferating hepatocyte stem/progenitor cells under feeder-free conditions without using cell-sorting techniques. Our system will contribute to drug discovery and tailor-made medicine with the aim of dispensing the safest drugs for each individual.
Materials and Methods
Generation and culture of human iPS cells
A human iPS cell line, 253G1, was established by transducing human adult skin fibroblasts with retrovirus containing Oct3/4, Sox2, Klf4, and/or c-Myc, as described previously (Takahashi et al., 2007). Human iPS cell line #40 was generated from human fetal lung fibroblasts (MRC-5 cells), via procedures described by Yamanaka and colleagues (Takahashi et al., 2007) with slight modifications (Nagata et al., 2009; Toyoda et al., 2011).
We also established human iPS cell line SeV5 without integration of viral vector components from human neonatal fibroblasts using Sendai virus (SeV) vectors, as described previously (Ban et al., 2011; Fusaki et al., 2009). Human fibroblast cell line BJ from neonatal foreskin (ATCC, Manassas, VA) were infected with SeV vectors containing Oct3/4, Sox2, Klf4, or c-Myc and were then incubated for 6 days in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (PAA Laboratories GmbH, Linz, Austria). Then the cells were cultured on dishes coated with γ-irradiated murine embryonic fibroblasts (MEFs) in Primate ES cell medium (ReproCELL Inc., Tokyo, Japan) for 3 weeks. Human ES cell-like colonies were picked up using a micropipette and were further cultured on dishes coated with γ-irradiated MEFs. These human ES-like cells expressed several multipotent markers such as SSEA4, Oct3/4, and Nanog. SeV and transgenes in human iPS cells were diluted to undetectable levels during repeated passage for approximately 2 months and/or were deleted by high-temperature cultivation (at 39°C) for 7 days (Ban et al., 2011).
All human iPS cells were maintained on dishes coated with γ-irradiated MEFs as described previously (Gokoh et al., 2011).
Culture of human ES cells and normal human hepatocyte cell lines
The use of human ES cells was performed in accordance with the Guidelines on the Utilization of Human Embryonic Stem Cells of the Ministry of Education, Culture, Sports, Science, and Technology of Japan, after approval by the institutional review board of the National Center for Global Health and Medicine. Human ES cells (KhES-1) (Suemori et al., 2006), provided by Kyoto University (Kyoto, Japan), were maintained as described previously (Nakahara et al., 2009a, 2009b; Saeki et al., 2009).
We used two human hepatic cell lines as positive control. HepG2 cell was purchased from Research Resources Bank of Japan Health Science Foundation (Osaka, Japan) and cultured in DMEM supplemented with 10% heat-inactivated FBS (PAA Laboratories). HepaRG cells (Gripon et al., 2002) was purchased from BIOPREDIC INTERNATIONAL (Rennes, France) and cultured on collagen IV-coated dish in General Purpose medium 670, Maintenance and Metabolism medium 620, or Induction medium 640 (BIOPREDIC INTERNATIONAL), according to the protocol of the supplier.
Hepatocyte differentiation of human iPS and ES cells in nonfeeder and serum-free culture
Before the induction of differentiation, human iPS and ES cells maintained on MEF were detached with dissociation liquid containing 0.25% trypsin (Invitrogen Corp., Carlsbad, CA), 1 mg/mL collagenase IV (WAKO Pure Chemical Industries, Osaka, Japan), 20% Knockout™ Serum Replacement (KSR) (Invitrogen), and 1 mM CaCl2. Detached cells were collected into conical tube, and were then allowed to stand at room temperature to sediment iPS/ES cells. After appropriate periods (approximately a few minutes), MEF in the supernatant of the tube were aspirated and iPS/ES cells at the bottom of the tube were collected. Collected human iPS/ES cells were then cultured for 2–3 days on matrigel-coated dish in DMEM/F12 medium supplemented with 20% KSR (Invitrogen), 1% nonessential amino acid solution (Invitrogen), 1 mM sodium pyruvate solution, 100 μM 2-mercaptethanol, 2 mM L-glutamine, 20 U/mL penicillin, and 20 μg/mL streptomycin.
The protocol for differentiation induction culture is summarized in Figure 1. Human iPS/ES cells were culture on matrigel-coated dishes in RPMI1640 medium supplemented with 2 mM L-glutamine, 100 μM 2-mercaptoethanol, 20 U/mL penicillin 20 μg/mL streptomycin, and 0.29% recombinant human albumin (Mitsubishi Tanabe Pharma Corp., Osaka, Japan) in the presence of 100 ng/mL Activin A (PeproTech, Rocky Hill, NJ), and 25 ng/mL Wnt 3A (R&D Systems Inc., Minneapolis, MN) for 24 h, followed by another 24-h culture in the presence of Activin A alone in the presence of 0.2% KSR (Invitrogen). After 48 h from the beginning of differentiation culture, the cells were subjected to the next stage of culture and were cultured with mixtures of three cytokines, fibroblast growth factor-2 (FGF-2; PeproTech; 10 ng/mL), bone morphogenic protein-4 (BMP-4; Wako Pure Chemical Industry; 20 ng/mL) and Sonic hedgehog (Shh; R&D; 200 ng/mL) in the presence of 2% KSR for 5 days. Then, the cells were culture with hepatocyte growth factor (HGF; R&D; 20 ng/mL), FGF-2 (10 ng/mL), and BMP-4 (20 ng/mL) for another 5 days in similar medium. Finally, cells were culture in the presence of oncostatin M (R&D; 10 ng/mL) and dexamethasone (0.1 μM) for final hepatic maturation for 6 to 16 days.
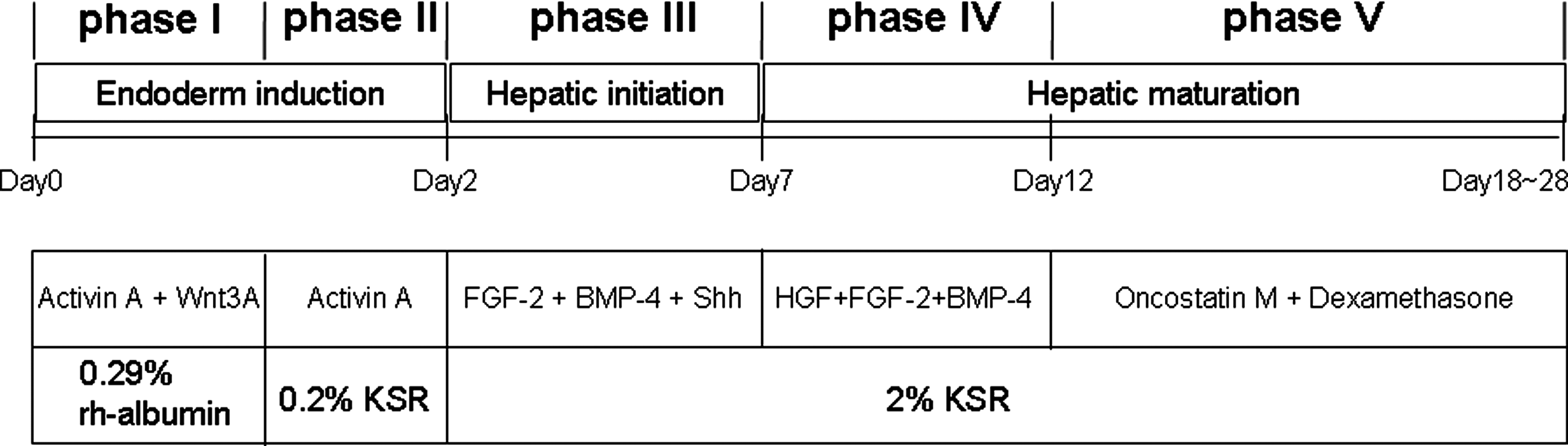
Schematic presentation of feeder-free and serum-free production of functional hepatocytes from human iPS and ES cells. Undifferentiated human iPS and ES cells were induced to differentiate into hepatocytes using a five phase culture system that mimics developmental process of liver in vivo. Abbreviations: FGF-2: fibroblast growth factor 2, BMP-4: bone morphogenic protein 4, Shh: Sonic hedgehog, HGF: hepatocyte growth factor, KSR: Knockout™ Serum Replacement, rh-albumin: recombinant human albumin.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from the cultured cells using TRIzol Reagent (Life Technologies co., Carlsbad, CA). For reverse transcription reactions, 1 μg RNA was reverse-transcribed using Superscript™ III First-Strand Synthesis System for RT-PCR (Invitrogen). A total of 0.5 μL of cDNA was used for PCR analysis. PCR amplification of different genes was performed using rTaq (Takara, Tokyo, Japan), with a program comprising 94°C for 5 min, 35 cycles of 94°C for 30 sec, 50–60°C for 30 sec, 72°C for 30 sec, and a final extension at 72°C for 10 min. The sequence of the primers used are as follows: α-fetoprotein (AFP); a forward primer 5′-TTTTGGGACCCGAACTTTCC-3′ and a reverse primer 5′-CTCCTGGTATCCTTTAGCAACTCT-3′, albumin (ALB); a forward primer 5′-GGTGTTGATTGCCTTTGCTC-3′ and a reverse primer 5′-CCCTTCATCCCGAAGTTCAT-3′, α1-antitrypsin (AAT); a forward primer 5′-ACATTTACCCAAACTGTCCATT-3′ and a reverse primer 5′-GCTTCAGTCCCTTTCTCGTC-3′, cytochrome P450 3A4 (CYP3A4); a forward primer 5′-ATGAAAGAAAGTCGCCTCG-3′ and a reverse primer 5′-TGGTGCCTTATTGGGTAA-3′, hepatocyte nuclear factor 4α (HNF-4α); a forward primer 5′-CCACGGGCAAACACTACGG-3′ and a reverse primer 5′-GGCAGGCTGCTGTCCTCAT-3′, tyrosine aminotransferase (TAT); a forward primer 5′-CCCCTGTGGGTCAGTGTT-3′ and a reverse primer 5′-GTGCGACATAGGATGCTTTT-3′, tryptophan 2, 3-dioxygenase (TDO2); a forward primer 5′-TACAGAGCACTTCAGGGAG-3′ and a reverse primer 5′-CTTCGGTATCCAGTGTCG-3′, glyceraldehyde 3 phosphate dehydrogenase (GADPH); a forward primer 5′-GAAGGTGAAGGTCGGAGTC-3′ and a reverse primer 5′- GAAGATGGTGATGGGATTTC-3′.
Quantitative RT-PCR
Total RNA was isolated by using TRIzol reagent and cDNA was synthesized in 20 μL of reaction volume containing 1 μg of total RNA and Superscript™ III First-Strand Synthesis System for RT-PCR (Invitrogen) in accordance with the manufacturer's instructions. Duplex real-time PCR (target gene and glyceraldehyde 3 phosphate dehydrogenase (GAPDH) as a reference gene) in 96-well optical plates was performed using TaqMan® technology and analyzed using an ABI PRISM® PE7900 HT sequence Detection System (Perkin-Elmer Applied Biosystems, Lincoln, CA). PCR mix per well (25 μL) consisted of commercially available, premixed GAPDH TaqMan® primers/probe, TaqMan® Gene Expression Assays, inventoried primers/probe for the target gene, 0.5 μL cDNA and QuantiTect® Multiplex PCR Master Mix (Qiagen, Valencia, CA). PCR conditions were as follows: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec, and 60°C for 1 min. The expression level of each gene was normalized to RNA content for each sample by using GAPDH as an internal control.
Western blotting
Western blotting was performed using rabbit polyclonal antihuman AFP (Epitomics Inc., Burlingame, CA), rabbit polyclonal antihuman ALB (DakoCytomation, Glostrup, Denmark), rabbit polyclonal antihuman AAT (Lifespan Bioscience Inc., Seattle, WA), and rabbit polyclonal anti-human CYP 3A4 antibodies (Abcam, Cambridge, UK). The second antibody reaction was performed using a horseradish peroxidase-conjugated antirabbit or antimouse IgG (Cell Signaling Technology, Inc., Beverly, MA). The final detection procedure was performed using ECL Western blotting detection reagents (GE Healthcare UK Ltd., Buckinghamshire, UK).
Immunostaining
The cells were fixed with acetone/methanol solution (1:3), permeabilized with 0.1% Triton X-100 in phosphate-buffered saline and blocked with BlockAce (DS Pharma Biomedical). The immunostaining procedure was performed with primary antibody reactions using a rabbit polyclonal antihuman AFP antibody (Epitomics), rabbit polyclonal antihuman AAT antibody (Lifespan Bioscience), rabbit polyclonal antihuman CYP3A4 (Abcam), mouse monoclonal antihuman epithelial cell adhesion molecule (EpCAM) antibody (Cell Signaling Technology), and a rabbit polyclonal antihuman ALB antibody (DakoCytomation), followed by secondary antibody reactions using Alexa Fluor® 488 chicken antimouse IgG (H+L), Alexa Fluor® 568 goat anti-rabbit IgG (H+L) or Alexa Fluor® 594 chicken antigoat IgG (H+L) (Invitrogen) antibodies.
Periodic Acid Schiff (PAS) assay for glycogen storage
Glycogen storage was measured by PAS staining a using a PAS staining kit (Muto Pure Chemicals, Tokyo, Japan) in accordance with the manufacturer's instructions.
Cellular uptake and release of Indocyanine Green (ICG)
ICG (Sigma-Aldrich, St. Louis, MO) was dissolved in DMSO to make a stock at 5 mg/mL and then freshly diluted in culture medium to 1 mg/mL. After incubation of cells with ICG (Sigma-Aldrich) for 30 min at 37°C, the medium with ICG was discarded and washed three times with phosphate-buffered saline, and the cellular uptake of ICG was examined by microscopy. Cells were then returned to the culture medium and incubated for 6 h for the release of cellular ICG stain.
Cytochrome P450 activity assay
CYP3A4 activity was evaluated using a p450-GloTM CYP3A4 Assay kit (Promega, Madison, WI). The cells were treated with or without dexamethasone (50 μM) for 16 h for induction and were then incubated with culture medium supplemented with 50 μM CYP3A4 substrates in accordance with the manufacturer's instructions. At 4 h after treatment, 50 μL of culture medium was removed and assayed in a luminometer. CYP450 activities were expressed as relative light units (RLU/mL).
Hepatocyte toxicity assay by D-galactosamine (D-GalN)
The cells were treated with 25 mM D-GalN for 24 h at 37°C, and then the supernatant was collected. Glutamic oxaloacetic transaminase (GOT), glutamic pyruvic transaminase (GPT), γ-glutamyl transpeptidase (γ-GTP), leucine aminopeptidase (LAP), and isozymes of lactate dehydrogenase (LDH) in the culture medium were measured sing a routine colorimetric laboratory method. In some experiments, cells were preincubated with 4 mM prostaglandin E1 (PGE1) for 4 h at 37°C before D-GalN was added.
Transmission electron microscopy
The samples were fixed using 2.5% glutaraldehyde in 0.1 M PBS (pH 7.4) for 2 h. After washing with PBS, samples were postfixed with 2% osmium tetraoxide for 1 h. Samples were dehydrated in a series of ascending ethanol concentrations and placed in propylene oxide prior to embedding in epoxy resin (Sakura Finetek Japan, Tokyo, Japan). After resin polymerization, sections of approximately 60∼80 nm were cut using Ultracuts (Reichert Scientific Instruments Co., Buffalo, NY) and double stained with uranyl acetate and lead citrate. Electron micrographs were taken using a Hitachi H-7500 transmission electron microscope (Hitachi High-Technologies Corp., Tokyo, Japan).
Cell proliferation assay
We used a Cellomics® BrdU and Ki-67 cell proliferation kit (Thermo Fisher Scientific Inc., Waltham, MA) and the assay was performed in accordance with the manufacturer's instructions. The cells were labeled with 120 μM BrdU for 20 h and then permeabilized and incubated with mouse anti-BrdU and rabbit anti-Ki-67 antibodies for 60 min at 37°C. After washing, the cells were incubated with Alexa Fluor 488-conjugated goat antimouse IgG, Alexa Fluor 549-conjugated goat antirabbit IgG and DAPI, washed twice, and mounted on glass slides with ProLong® Gold antifade reagent (Invitrogen). The slides were subsequently inspected using a fluorescence microscope.
Experiments for passages and regrowth of induced hepatocytes
At the 28th day of differentiation, induced hepatocytes were collected from the original culture dishes using a StemPro® EZPassage™ Disposable Stem Cell Passaging Tool (Invitrogen) and were recultured in new 3.5 mmφcollagen-coated dishes (ASAHI GLASS Co., Ltd., Tokyo, Japan).
Results
Feeder-free and serum-free culture methods for hepatocyte differentiation of human iPS and ES cells
After a process of trial and error, we found that the key to success in inducing endodermal differentiation of human ES/iPS cells resided in preparing the starting materials at appropriate densities, that is, to seed the undifferentiated human ES/iPS cells so that the clumps of cells were just contacting one another. By using these appropriately seeded cells, we successfully performed a three-stage (five-phase) differentiation protocol for the induction of functional hepatocytes from human iPS and ES cells (Fig. 1).
In the initial experiment, we performed differentiation induction of human iPS cells (253G1) established from adult human dermal fibroblasts using retrovirus vectors, because this cell line is one of the standard iPS cell lines in Japan and was established without using a protooncogene c-Myc. During differentiation induction, we analyzed time courses of expression of hepatocyte-specific genes (AFP, ALB, and hepatocyte-specific metabolic enzymes). As shown in Figure 2A, transcriptional expression of hepatocytes-specific markers such as AFP, ALB, AAT, tyrosine aminotransferase (TAT), CYP3A4, and tryptophan 2, 3-dioxygenease (TDO2) were observed during differentiation, although the expression levels of TAT, CYP3A4, and TDO2 were slightly weaker than the other markers. To evaluate more quantitatively, we then performed quantitative real time PCR studies of these three markers, TAT, CYP3A4, and TDO2 and compared them with those of human hepatocyte cell lines, HepaRG and HepG2. As shown in Figure 2B, cellular expression of these three markers was detected in a time-dependent manner and the level of their expression was quantitatively equivalent to or sometimes much better than the level of human hepatocyte cell lines.
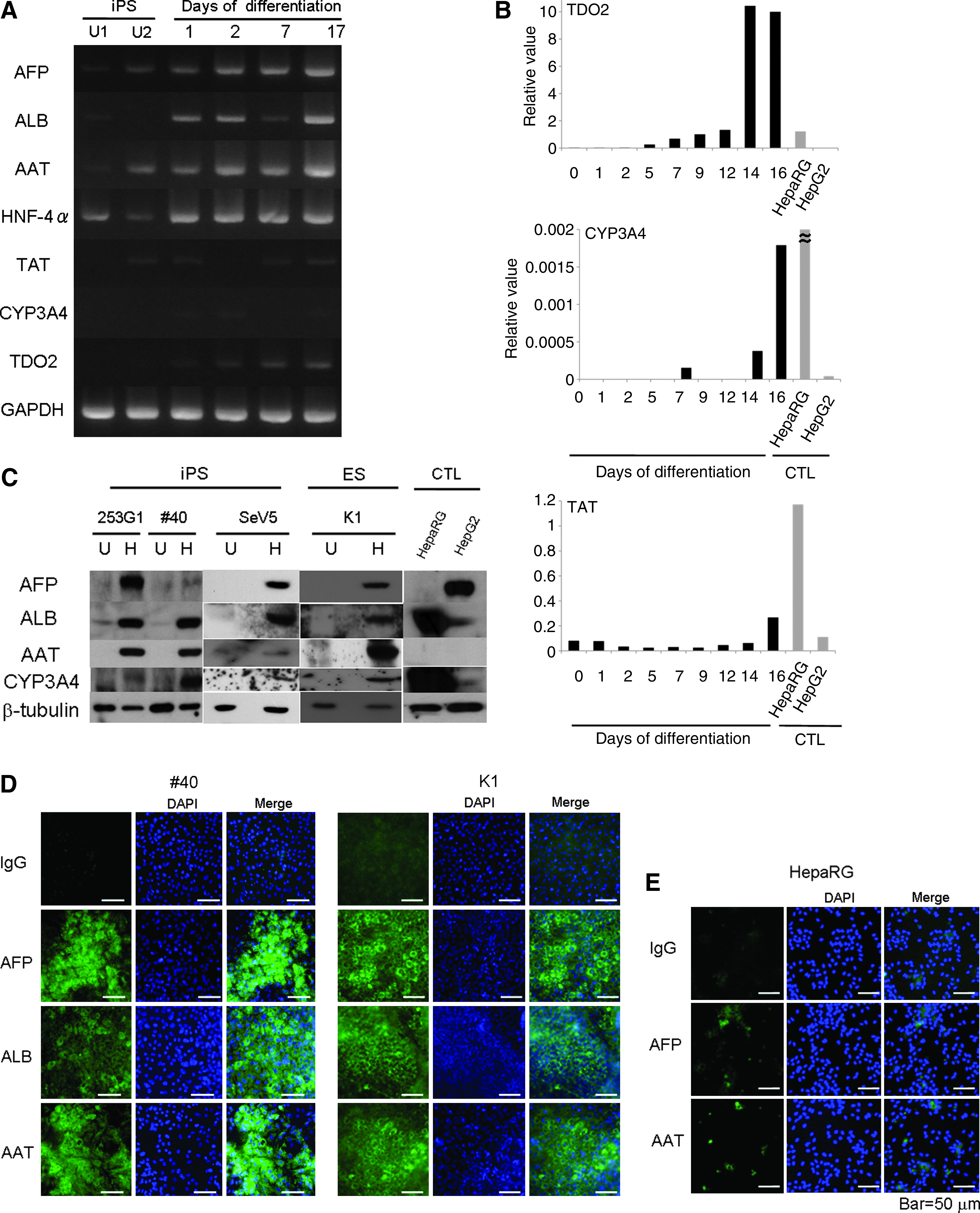
Molecular characterization of hepatocytes induced from human iPS and ES cells. (
We then confirmed the expression of hepatocyte markers at the protein level. In these experiments, we also used two other human iPS cell lines, one was #40 established from fetal fibroblasts using a retrovirus vector, and the other was SeV5 established from neonatal fibroblasts using Sendai virus vector, without integration of viral vector components and transgenes. As shown in Figure 2C, several hepatocyte markers, such as AFP, ALB, AAT, and CYP3A4, were induced in all three human iPS cells. The level of induction was similar to that of human hepatocyte cell lines. In particular, AAT, which was not detected in control human hepatocyte cell lines, was prominently induced in human iPS cells. These findings were further confirmed using human ES cells (KhES-1) (Fig. 2C). Protein expression of AFP, ALB, and AAT was also confirmed by immunostaining (Fig. 2D), and overall positivity for these three markers was approximately 30–60%. In contrast, protein expression of AFP and AAT in HepaRG cells as determined by immunostaining was weak and positivity was less than 20% (Fig. 2E).
Functional evaluation of hepatocytes induced from human iPS and ES cells
To clarify whether human hepatocytes induced from human pluripotent stem cells possessed the functional capacity of mature hepatocytes, we performed three standard assays, ICG-uptake capacity, glycogen storage capacity, and CYP450 activity. As shown in Figure 3A, hepatocytes differentiated from human iPS cells (253G1, #40, and SeV5) and human ES cells (KhES-1) all showed clear uptake of ICG, and this ICG was released after 6 h. The overall level of ICG uptake-positive cells was approximately 20–30%. HepaRG and HepG2 cells showed substantial capacity (20–30% for HepaRG cells and less than 5% for HepG2 cells). Thus, hepatocytes from human iPS and ES cells have sufficiently mature functions for hepatocytes compared with the levels in control hepatocytes.

Functional activity of hepatocytes induced from human iPS and ES cells. (
Then, we evaluated cytoplasmic glycogen accumulation in undifferentiated human iPS and ES cells and hepatocytes induced from these pluripotent stem cells, and found that almost all (more than 80%) of hepatocytes induced from human iPS and ES cells were strongly positive for PAS staining, whereas undifferentiated human iPS and ES cells were not stained (Fig. 3B). On the other hand, HepaRG cells were almost 50–60% positive, and Hep G2 cells were negative.
It has been reported that CYP3A4 plays a central role in drug metabolism and detoxification in the liver among a subfamily of CYP450s (Liu et al., 2007), and we identified the expression of this critical enzyme during our differentiation culture at both mRNA (Fig. 1) and protein (Fig. 2) levels. We then determined the functional activity of CYP3A4 using hepatocytes differentiated from human iPS and ES cells. As shown in Figure 3C, hepatocytes induced from human iPS and ES cells showed high levels of CYP3A4 activity compared with those of undifferentiated human iPS and ES cells and human hepatic cell lines, HepaRG and HepG2 cells. In addition, high CYP3A4 activity in the differentiated cells was further potentiated by transient (16 h) pharmacological induction in hepatocytes induced from human iPS cells (#40) and human ES cells (KhES-1).
Hepatocyte toxicity assay using D-galactosamine
It is essential to establish an in vitro hepatocyte cytotoxic assay for the evaluation of liver toxicity of various substance and/or drugs. Therefore, we performed an in vitro cytotoxic assay with hepatocytes induced from human pluripotent stem cells using a traditional method with D-GalN (Bao and Liu, 2010; Kuhla et al., 2009; Siendones et al., 2005). GOT, GPT, γ-GTP, LAP, and LDH isozymes released from the cells were quantified as an indicator of hepatocyte-specific cytotoxicity.
As shown in Figure 4A, D-GalN significantly and potently induced the extracellular release of GOT, GPT, and/or LDH into the culture medium, and the pattern of LDH was hepatocyte-specific (LDH4/5 dominant) in hepatocytes induced from human iPS and ES cells. These cytotoxic effects of D-GalN were significantly inhibited by PGE1 (Fig. 4B) (Siendones et al., 2005). In contrast, D-GalN only minimally induced the extracellular release of these hepatocyte-specific enzymes in undifferentiated human iPS and ES cells and human umbilical vein endothelial cells (HUVECs). Extracellular release of GOT, GPT, and LDH5 into the medium was also observed in control hepatocytes, HepaRG and HepG2 cells. Interestingly and unexpectedly, γ-GTP and LAP, which are bile duct specific enzymes, were released from “hepatocytes” induced from human iPS and ES cells but not undifferentiated human iPS and ES cells, HUVEC, and control hepatocytes (HepaRG and HepG2 cells). These findings suggest that “hepatocytes” induced from human iPS and ES cells include not only hepatocytes themselves but also bile duct-related cells such as cholangiocytes. Thus, our culture system could be useful for cytotoxicity assays for both hepatocytes and cholangiocytes.

D-galactosamine cytotoxic assay. (
Electron microscopic study of hepatocytes and cholangiocytes in our culture system
To confirm that the “hepatocytes” induced from human pluripotent stem cells were really hepatocytes and explore possible coexistence of bile ducts, bile canaliculi, and cholangiocytes in the culture system, we performed detailed morphological examinations of differentiated cells using an electron microscope.
As shown in Figure 5A, hepatocytes induced from human ES cells were equipped with microvilli of intermediate length (shorter than the microvilli in the intestine) on their open space-side [“space of Disse” (perisinusoidal space) in the liver] and adjacent cells were connected via structures such as tight junctions and desmosomes. Between the cells, there was a microduct-like structure with microvilli on the lumen side, which was considered to be bile canaliculus, and this microduct-like structure was also joined by cell–cell connecting structures (tight junctions and desmosomes). Abundant glycogen α-particles were observed in the cytoplasm, and these glycogen α-particles showed a hepatocyte-specific “rosette formation.” We also observed “peroxisomes,” cytoplasmic structures highly specific for hepatocytes. Similar microscopic findings were observed in hepatocyte-like cells induced from human iPS cells (Fig. 5B). In addition, typical structures specific for bile ducts were also observed, that is, bile duct epithelial cells with short microvilli on the lumen side and the basement membrane on the other side were observed (Fig. 5C).

Transmission electron micrographs of hepatocyte and cholangiocytes induced from human ES and iPS cells. (
Thus, there existed at least three cell types and structures, hepatocytes, bile canaliculi (intrahepatic microbile duct) and bile duct epithelial cells, and all of these microstructures are extremely specific for the liver (Ghadially, 1997).
Possible existence of bipotential hepatoblasts with proliferating capacity
Data presented in Figures 4 and 5 together clearly indicate the presence of both hepatocytes and cholangiocytes in the present culture system. It is has been proposed that both hepatocytes and cholangiocytes are derived from their common progenitor cells called hepatoblasts (Zhao and Duncan, 2005). We then explored the presence of immature progenitors with proliferating potential.
Figure 6A shows cell morphologies observed using an inverted microscope. Two forms of cell structures and/or areas were observed: one was a bulging cell clump area, and the other was a flattened monolayer area containing binuclear cells. These two areas were analyzed using immunostaining for AFP and EpCAM, both of which are hepatoblasts markers (Schmelzer et al., 2006). As shown in Figure 6B, the bulging areas were stained positively for both markers, whereas the flattened areas were not. These findings suggested that the present culture system mainly contained two populations: one in the bulging area with proliferating immature cells with hepatoblast markers, and the other in the flattened mature hepatocyte area. The former area was also positive for proliferation marker Ki-67 (Fig. 6C), and the latter areas were also positive for ALB, a mature hepatocyte marker (Fig. 6D). In addition, we confirmed these findings in a single microscopic field; namely, the bulging area was positive for BrdU, a proliferation marker, and the flattened area was positive for AAT, a mature hepatocyte marker (Fig. 6E). Thus, the bulging area with frequent cell nuclei (stained by DAPI) was positive for proliferation markers (Ki-67 and BrdU) and hepatoblast markers (AFP and EpCAM), whereas the flattened area had fewer cell nuclei (stained by DAPI) and was negative or weakly positive for proliferating markers and positive for mature hepatocyte markers (ALB and AAT).
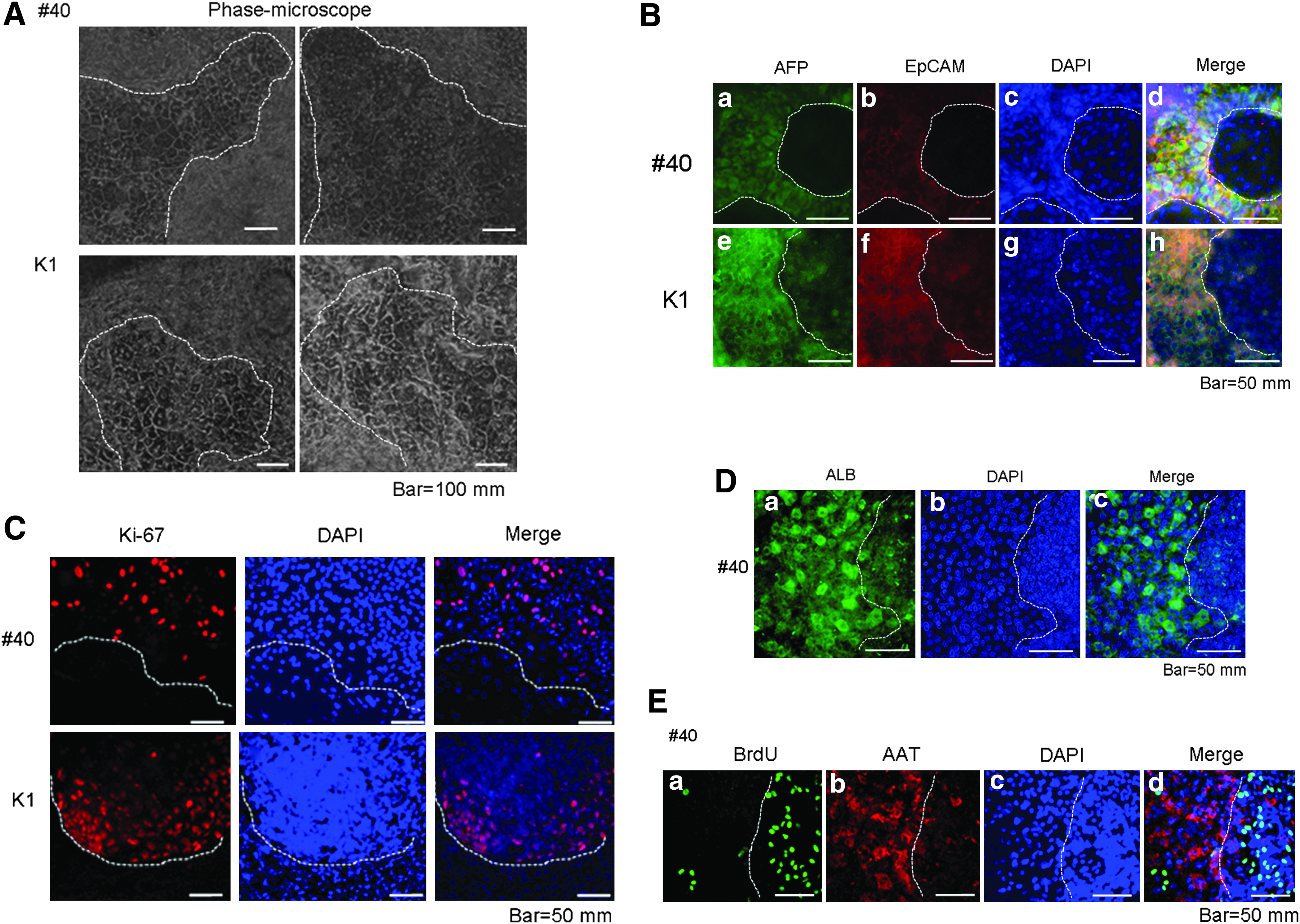
Immature proliferating and mature hepatocyte fractions in induced cells. (
To further confirm the proliferative potential of “hepatocytes” from human pluripotent stem cells, we performed passages of differentiated cells into new culture dishes. Induced hepatocytes were collected from the original culture dish and recultured in new dishes. After attaching to the new dish, cells began to proliferate again (Fig. 6F, upper panel). On the second day of reculture, colonies of small-sized cells (10–20 μm vs. 20–60 μm; cells of flattened areas in Fig. 6A) appeared and expanded during reculture for up to 9 days (Fig. 6F, upper panel). These small-sized cells were positive for AFP and AAT (Fig. 6F, lower panel).
Thus our differentiation culture included not only mature hepatocytes but also their precursors with proliferation potential and began to proliferate again even after transfer to new culture dishes along with positive results for hepatocyte markers.
Discussion
In the present study, we established a feeder-free and serum-free induction system for human mature, functional hepatocytes from human pluripotent stem cells, iPS, and ES cells. We found that an initial high density culture, so that each colony of undifferentiated human ES/iPS cells was contacting other colonies, was essential in order to start stable and reproducible differentiation. In addition, we used a high concentration of Activin A and added Wint 3A to induce effective initial endoderm differentiation (Hay et al., 2008a). We also added Shh during second-step differentiation because Hedgehog has been reported to play important roles during liver development in embryos (Hirose et al., 2009) and liver regeneration after hepatectomy (Ochoa et al., 2010), despite the fact that there are no reports to use Hedgehog for hepatic differentiation of human ES/iPS cells.
There have been many reports to show in vitro differentiation of human ES/iPS cells toward hepatocytes, although most of the studies utilizes animal-derived cells and/or materials during at least one step of differentiation cultures (Agarwal et al., 2008; Cai et al., 2007; Chiao et al., 2008; Duan et al., 2007, 2010; Hay et al., 2008a, 2008b; Inamura et al., 2011; Ishii et al., 2008; Liu et al., 2010; Mfopou et al., 2010; Sasaki et al., 2009; Si-Tayeb et al., 2010; Song et al., 2009; Sullivan et al., 2010; Touboul et al., 2010; Zhao et al., 2009), such as the presence of MEF or MEF-conditioned medium at the initial step of the culture, the presence of mouse feeder cells for differentiation induction, or fetal bovine serum during the certain periods of differentiation culture. In contrast, the only xenogeneic materials used in our differentiation protocol were Matrigel and KSR, both of which may be substituted by equivalent products of human origin. The present results contribute to the development of production techniques for human hepatocytes free from animal products and a good product level.
In this study, we succeeded in inducing functional hepatocytes from several human pluripotent stem cells with distinct origins and diverse characteristics. We used human iPS cells from adult fibroblast, neonatal fibroblasts, and fetal fibroblasts and in addition human ES cells. Induction methods of iPS cells also varied, that is, traditional retrovirus vector and Sendai virus vector. Despite these variations, we were able to reproduce stable and effective differentiation in all situations. Thus, our differentiation-inducing method is universal and might be widely applicable.
Human iPS cells were originally been established using retrovirus and lentivirus vectors (Park et al., 2008; Takahashi and Yamanaka, 2006; Takahashi et al., 2007; Yu et al., 2007), and these methods for iPS induction have been of major concern for the clinical application of iPS cells. On the other hand, RNA-based methods (Warren et al., 2010), including SeV vectors (Ban et al., 2011; Fusaki et al., 2009) are really hopeful and lead to a reproducible supply of safe human iPS cells. In this study, using Sendai virus vectors and our efficient differentiation methods, we succeeded in genome virus free production of human mature hepatocyte from safe human iPS cells.
We demonstrated sufficient induction of CYP3A4 in differentiated hepatocytes at the levels of mRNA, protein and activity. It is well known that many drugs are metabolized by CYP3A4 and many drugs inhibit its activity, and furthermore several drugs induce its expression (Liu et al., 2007). In this study, we observed an additional induction of CYP3A4 by dexamethasone. These findings suggest the physiological functionality of hepatocytes derived from human pluripotent stem cells in the present study and usefulness of these differentiated cells as in vitro drug metabolic studies.
In addition to CYP3A4, we demonstrated the induction of AAT at the protein level in human hepatocytes induced from human pluripotent stem cells, and this protein was not detected in control human hepatocytes used as positive control. In this point, therefore, human hepatocytes derived from human ES/iPS cells via our culture system are superior to the commercial human hepatocytes used so far. AAT is an inhibitor for proteinase and is thought to play an important role in regulating a wide variety of proteolytic reactions in the human body. Congenital deficiency of this enzyme is associated with serious lung and liver damage, and Rashid et al. (2010) reported the establishment of patient-specific iPS cells of this enzyme deficiency. Thus, our culture system may contribute to the analysis of this congenital disease. In contrast to CYP3A4, AAT is released from hepatocytes into the bloodstream after being produced. Thus, we believe that human hepatocytes generated using the present culture protocol have full capacity to produce two important functional proteins working inside and outside the liver, and thereby contributing homeostasis in the human body.
In the present study, we clarified that hepatocytes differentiated from human pluripotent stem cells could be useful for in vitro hepatocyte cytotoxicity assays. Unlike the results of previous studies that showed an in vitro apoptosis-related phenomenon (Bao et al., 2010) and extracellular release of total LDH (Siendones et al., 2005) and/or performed in in vivo animal studies (Kuhla et al., 2009), we identified the extracellular release of hepatocyte-specific enzymes, such as GOT and hepatocyte-specific isozymes of LDH, using in vitro human culture systems. In addition, we also showed that the cytotoxicity was really specific for hepatocytes because cytotoxicity was not or only minimally observed in the cells other than hepatocytes including undifferentiated human pluripotent stem cells and HUVEC. These results are the first demonstration of hepatocyte-specific cytotoxicity particularly in induced cells derived from human pluripotent stem cells, and the convenient in vitro assay system might be applicable to various in vitro testing approaches for substances that are toxic to the liver. In addition, our cytotoxic assay might also be used for cytotoxicity assays for epithelial cells of the biliary tract, because extracellular release of γ-GTP and LAP were detected upon cytotoxic stimulation. This toxicity of hepatocytes (and probably biliary epithelial cells) was attenuated by hepatoprotective PGE1, showing a possible therapeutic model of human liver damage, because PGE1 is a candidate hepatoprotective agent also in the clinical settings (Hara et al., 2010).
In our differentiation-inducing culture system, not only hepatocytes but also cholangiocytes seems to be induced concomitantly, as suggested by the cytotoxic study described above. These findings were confirmed by morphological studies using electron microscopy. It is well known that both hepatocytes and cholangiocytes are derived from common precursor cells called “hepatoblasts” (Zhao and Duncan, 2005). Because the differentiated cell in our culture system expressed AFP, it is likely that our culture system contains hepatoblasts even at the terminal stage of differentiation. In the present study, we identified AFP and EpCAM double-positive cells in our culture system and these cells were also positive for Ki-67 and BrdU, suggesting the presence of hepatocyte progenitors with high proliferative potential. We also showed that induced cells were passageable and did proliferate again in new dishes. In addition, proliferating cells (colony-forming small-sized cells) in the new dish continued to express hepatocyte markers. These smaller hepatocytes with proliferating potential might be a human counterpart of rat small hepatocyte progenitor cells (Chen et al., 2007; Mitaka et al., 1992, 1995). We propose a possible in vivo functional reconstitution of liver with a bile acid delivering system to the bloodstream by transplanting simultaneously hepatocyte and cholangiocytes together with endothelial cells induced from human pluripotent stem cells, because we also succeeded in highly efficient endothelial induction from human ES/iPS cells (Gokoh et al., 2011; Nakahara et al., 2009a).
In this study, we used HepaRG cells as one of the control hepatocyte. This cell line has originally been established from hepatocytes in patients with type C hepatitis, but was free of hepatitis virus. In addition, HepaRG cells express a large panel of liver-specific genes, including those expressing drug metabolizing enzymes and therefore were widely used as human hepatocyte model in many recent reports (Guguen-Guillouzo and Guillouzo, 2010; Marion et al. 2010). Despite this, there are no reports with this cell line of hepatocyte induction from human ES/iPS cells. We could show that hepatocyte-like cells induced from human pluripotent stem cells via our culture system were almost equivalent or potentially superior to this standard hepatocyte.
In the present study, we presented novel efficient method to produce functional hepatocytes from pluripotent stem cells. In regard to the possible methods for hepatocyte production, there is another important option, namely, direct reprogramming from somatic cells. Recent two reports (Huang et al., 2011; Sekiya and Suzuki, 2011) clearly showed the usefulness of this strategy, and proposed several defined factors for direct induction of hepatocytes from fibroblasts, which may be also useful for differentiation induction of hepatocytes from pluripotent stem cells. However, mouse cells and retrovirus and/or lentivirus vectors were used in these two reports. Further studies using human cells and more safe vectors are necessary for the clinical application of this powerful method.
Footnotes
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.