
Abstract
Abstract
Human embryonic stem cells maintained on human amniotic epithelial cells (hESCshAEC) are better preserved in an undifferentiated state and express pluripotency genes Oct4, Nanog, and Sox2 at higher levels compared with growth on mitotically inactivated mouse embryonic fibroblasts (hESCsMEF). Here we report that this correlates with the absence of the tumor suppressor and metabolic balancer gene, LKB1 expression in hESCshAEC. RNA interference knockdown of LKB1 in hESCsMEF resulted in upregulation of pluripotency marker genes of Oct4 and Nanog, while downregulation of differentiation markers (Runx1, AFP, GATA, Brachyury, Sox17 and Nestin). As in somatic cells, LKB1 controls p21/WAF1 expression by promoter binding in hESCsMEF. Our results suggested that the absence of LKB1-mediated signaling is an important determinant of feeder cell-mediated support of hESC renewal.
Introduction
Despite the similar origin, hESCs are distinct from naive mESCs, but more akin to mEpiSCs in morphology, developing potency, gene expression, and epigenetic modifications (Buecker et al., 2010). Consequently, hESCs have been considered to be developmentally more advanced than mESCs. This view has been challenged by recent report that hESCs bearing two active X chromosomes could be established from human blastocysts under physiological oxygen concentrations (Lengner et al., 2010). mESC-like hESCs have been achieved through gene manipulation combined with canonic mESCs culturing condition (Hanna et al., 2010). These studies suggest that hESCs could be as immature as mESCs, and the environmental conditions and culture methods might contribute to establish and maintain the naïve pluripotent state. However, the mechanisms by which such a transition is controlled remain to be elucidated.
In the quest to use hESCs as a source of cells for therapeutic application in regenerative medicine, it is important to develop humanized and defined culture environments supporting derivation, expansion, and differentiation free of animal-derived reagents, a potential source of adventitious pathogens. To this end we have previously established an optimized culture conditions to support the growth of undifferentiated hESCs, using primary human amnion epithelial cells (hAECs) as feeder cells, as an alternative to the traditional use of mitotically inactivated mouse embryonic fibroblasts (MEF) (Lai et al., 2010). By this method, we observed that the colony morphology of hESCs maintained on hAECs was domed similar to that of mESCs instead of traditional flattened morphology. Further, the expression of stemness markers such as Oct4, Nanog, and Sox2 in hESCs on hAECs was higher than hESCs maintained on MEF. hESCs grown on hAECs also showed distinct H3 acetylation and H3-K4 methylation status between Nanog and Oct-4 loci compared with hESCs on MEFs (Lai et al., 2010). However, the mechanism responsible for these differences was unknown.
To investigate the differences between hESCs supported by hAEC versus MEFs as a potential model system to understand mechanistic control of undifferentiated plutipotent states, we focused on a role for LKB1 (or serine/threonine kinase 11 - STK11), an evolutionarily conserved kinase, which has multiple cellular functions as a tumor suppressor and metabolic balance setting (Hemminki et al., 1998; Tiainen et al., 1999). LKB1 is essential for embryonic development in mice (Miyoshi et al., 2002). It is ubbiquitously expressed between E7 to E11 (Luukko et al., 1999), and Lkb1−/− mouse embryos fail in uterus between 8.5 and 9.5 days postcoitum (Jishage et al., 2002). In adult hematopoietic stem cells, LKB1 is necessary to maintain quiescence and metabolic activity (Gan et al., 2010; Gurumurthy et al., 2010; Nakada et al., 2010). Although LKB1 is important in regulating metabolic activity in cancer cells and some stem cells, its role on human ESC cells has not be explored. In the present study we implicate a role for LKB1 and its downstream targets p21/WAF1 (Tiainen et al., 1999; Zeng and Berger, 2006) in accounting for differences between hAEC versus MEF mediate supported of hESCs. We propose these pathways may be significant in the control of pluripotent cell states.
Materials and Methods
Cell culture
All procedures involving human subject research and ethics were approved by Shanghai Jiaotong University committees. The human ES cell line hHES1 (Wu et al., 2005) was presented by Dr. Wenjie Zhang. Human placentas were obtained after uncomplicated selective cesarean section from healthy mothers with written and informed consent. Amnion membranes were separated from the chorines membrane by mechanic peeling. Briefly, the amnion membrane were washed with 1×phosphate-buffered saline (PBS), then cut to yield 0.5–1.0 cm2 segments and digested with 0.25% trypsin/EDTA at 37°C for 45 min. The resulting cell suspension was cultured in RPMI 1640 medium supplemented with 10% KO serum replacement (Gibco, Grand Island, NY), streptomycin (100 U/mL), penicillin (100 U/mL), and glutamine (0.3 mg/mL). The growing hAECs reached to 70–80% confluence were used as a feeder layer for hESC culture.
The human ES cells were grown on MEFs or hAECs, where the culture medium consisted of KO-DMEM with 10% KO serum replacement, 2 mM L-glutamine, 0.1 mM nonessential amino acids, 0.1 mM 2-β-mercaptoethanol, 10 ng/mL bFGF, 12 ng/mL hLIF, and penicillin (25 U/mL)–streptomycin (925 mg/mL) mixture as previously described (Lai et al., 2009, 2010).
Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde in PBS and immunostained according to reported protocols using the primary antibodies against following proteins: Oct-4 (rabbit antihuman 1:500, Chemicon, Tumecula, CA), Nanog (rabbit antihuman 1:500, Chemicon), LKB1 (rabbit antihuman 1:500, Santa Cruz Biotechnology, Santa Cruz, CA), and P21 (rabbit antihuman 1:500, Santa Cruz Biotechnology). The antigen–antibody complex was labeled by 1:200 FITC-conjugated goat–antirabbit antibodies (Invitrogen, Carlsbad, CA). Fluorescence images were taken by a Leica MDI3000 microscope. DAPI (Roche, Indianapolis, IN) was used for nuclear staining as a reference (Lai et al., 2009, 2010).
RNA interference
LKB1-siRNA (CUGGUGGAUGUGUUAUACA) was used to knock down LKB1 in hESCs, and mock-siRNA (CACCAGCAUCUGAUCUAGA) was used as a negative control (Zeng et al., 2006). Transfection was done with lipofectamine (Invitrogen) at hESCs grown on MEF and hESCs on hAEC in 10-cm plates. siRNA oligos (1.0 nmol) were mixed with lipofectamine in Opti-MEM medium for 4–6 h. The culture media were replaced and hESCs were culturing for another 24 h.
Western blot
Total protein was determined in harvested cells using a BCA kit (Pierce, Gaithersburg, MD). Proteins were separated by SDS-polyacrylamide gel, and then electrophoresis was performed and transferred to nitrocellulose membrane, using antibodies specific to the target protein (Lai et al., 2009). Briefly, the nitrocellulose was incubated with the primary antibody against LKB1 (Santa Cruz Biotechnology, 1:500), P21 (Santa Cruz Biotechnology, 1:500), or β-actin (Cell Signaling, Beverely, MA; 1:1000). After an overnight incubation at room temperature, the nitrocellulose were washed thoroughly, and incubated with peroxidase-linked goat antirabbit-IgG (1:1000, Santa Cruz Biotechnology) at room temperature for 1 h. Chemiluminescence was performed using the Western lightning ECL kit (Perkin–Elmer Life Science, Norwalk, CT) and the ChemiImager imaging system (G: BOX SYNGENE, Gene Company Limited, Hong Kong).
Chromatin immunoprecipitation (ChIP) assays
The ChIP assays were carried out using antibodies against LKB1 (Santa Cruz Biotechnology, 1:500). Normal rabbit IgG (Catalog no. 12-370; Upstate, Lake Placid, NY) was used as a negative control to verify immunoprecipitation specificity. Briefly, cells were fixeded with 1% formaldehyde for 10 min at room temperature, then sonicated on ice and subjected to immunoprecipitation by incubating with antibodies at 48 oC overnight. Then the protein–DNA crosslinks are reversed and the DNA is purified and analyzed by PCR amplification under the following conditions: 95°C for 10 min, 32 cycles of 94°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec, final extension 72°C for 10 min (Primer sequences were shown on Table 1).
Electrophoretic mobility shift assay (EMSA)
Nonradioactive label EMSA was performed using Gel Shift Assay System (Promega, Madison, WI) according to the manufacturer's instructions. Five micrograms of nuclear extracts of hESCs on MEFs or hESCs on hAECs were incubated with DIG-labeled mutated probes in the reaction buffer for 20 min at room temperature. In competition experiment, prior to mixing labeled probe with nuclear extracts, the extracts were incubated with unlabeled oligonucleotides for 15 min at room temperature in the presence of 100-fold molar excess of labeled probe. The gel supershift assay was carried out by adding 2 μg of rabbit polyclonal anti-LKB1 antibody (Santa Cruz Biotechnology) to the DNA protein complex for 30 min at room temperature. All reaction samples were analyzed by the electrophoresis in 4% nondenatured polyacrylamide gels and exposed to Kodak MS X-ray films at −80°C. Oligonucleotides (sequences are shown in Table 1) were designed according to the cloned p21 promoter. Double-stranded Oligonucleotides were labeled by 5′-end-labeled with DIG DNA Labeling Mix (Roche cat #1277065).
Quantitative real-time PCR assay
Total RNA was extracted with Trizol and cDNA was synthesized by iScript cDNA synthesis kit (Bio-Rad, Hercules, CA) with 500 ng DNaseI treated RNA. The cDNA served as a template for the amplification of genes of interest and the housekeeping gene (18s mRNA) by real-time PCR, using IQ SYBR Green (Bio-Rad) and Mastercycler ep realplex Real-time PCR system (Germany).
Statistical analysis
Experimental data are expressed as mean±SD and the significance of differences was assessed with Student's t-test.
Results
LKB1 is specifically expressed in hESCsMEF
As previously reported, undifferentiated hESCS grown on primary human amnion epithelial cell (hESCshAEC) feeders have a distinctive domed morphology as distinct from a flattened morphology grown on MEF (hESCsMEF; Fig. 1A and B). This correlates with increased expression of pluripotency gene (Oct4 and Nanog) (Fig. 1C and D) and alterations in the epigenetic status of Nanog and Oct-4 promoters (Lai et al., 2010).
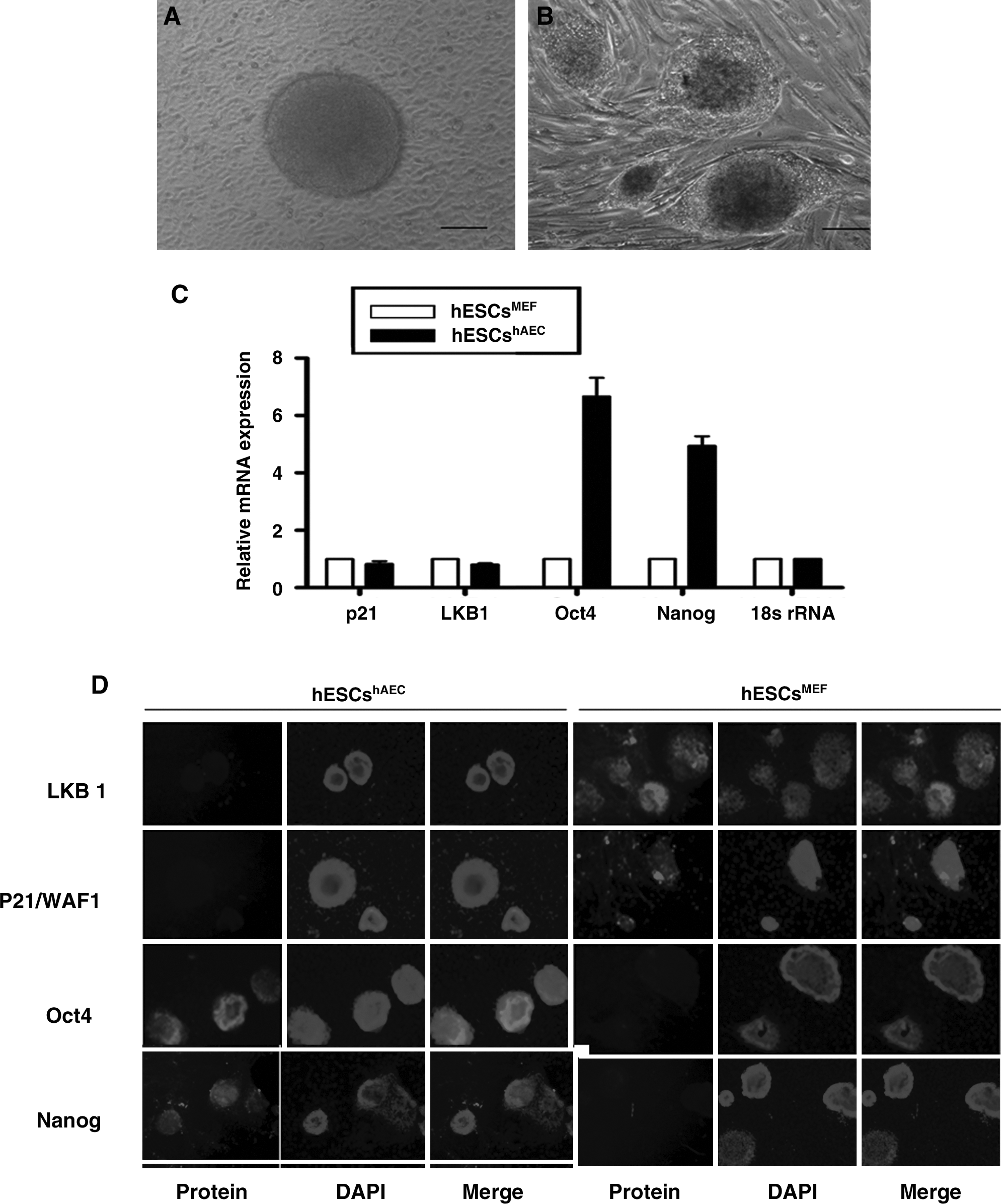
Human ESCs have distinct expression in different culture conditions. (
To study the putative role of LKB1 to account for these differences, we compared its expression in hESCshAEC and hESCsMEF. LKB1 mRNA and protein could not be detected in hESCshAEC through immunostaining, while the staining in hESCsMEF was clear. No staining for LKB1 was evident in either feeder cell population (data not shown). The same was observed for p21/WAF1, which mediates LKB1's tumor suppression function (Fig. 1C and D).
LKB1 directly regulates p21/WAF1 gene through promoter-binding in hESCs
LKB1's tumor suppressor effect is mediated by binding to the p21/WAF1 gene promoter and deletion of LKB1 gene increased the development of solid tumors (Gurumurthy et al., 2008). To study whether p21/WAF1 was the target of LKB1 in hESCs, we attempted to knock down LKB1 by RNAi and then evaluate the consequences to p21/WAF1 expression and promoter binding. LKB1-specific RNAi reduced LKB1 and p21/WAF1 expression in hESCsMEF as determined by Western blotting (Fig. 2A). Treatment with mock siRNA did not modify either LKB1 or p21/WAF1 expression. Neither protein was detected in hESCshAEC either before or after RNAi.
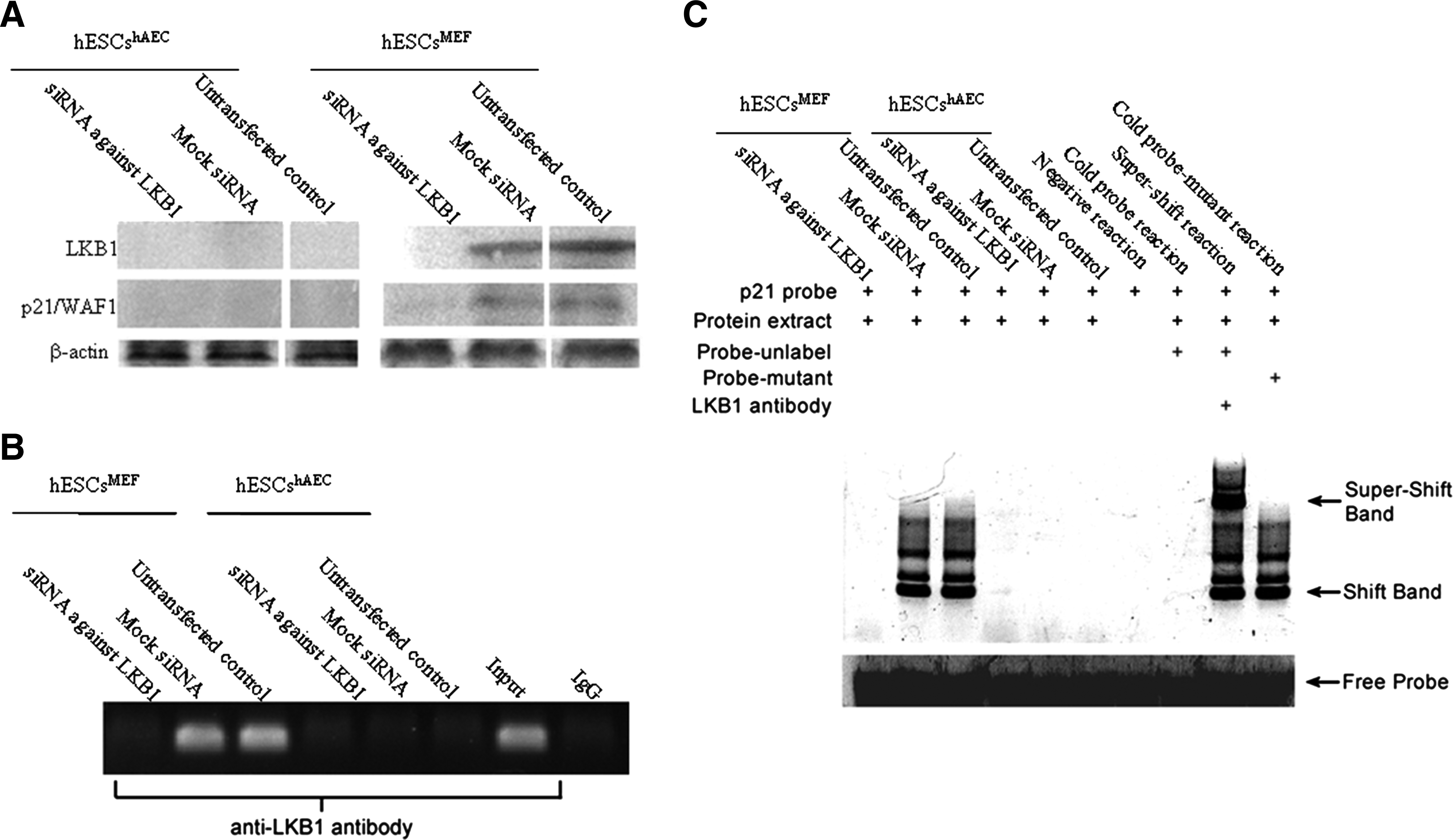
LKB1 directly regulates p21/WAF1 through promoter-binding in hESCs grown on MEF. (
As it has been reported that p53-dependent recruitment of LKB1 is associated to the promoter p21/WAF1 gene and has a direct role to mediate transcriptional activation in tumor cells (Zeng et al., 2006), we used chromatin immunoprecipitation assay to study whether it was also the case in hESCs. DNA segments corresponding to p21/WAF1 promoter region were amplified by PCR taking extracted DNA samples as templates before and after immunoprecipitation with an antibody to LKB1. As an input control, genome DNA of hESCshAEC was used as a template to amplify the p21/WAF1 promoter region. After immunoprecipitation against LKB1, no amplification existed in hESCshAEC, even in the untransfected control (Fig. 2B). This was not surprising, as the cellular concentration of LKB1 in hESCshAEC was quite low if there was any (Fig. 1 and Fig. 2A). In untransfected and mock siRNA transfected hESCsMEF, p21/WAF1 promoter region could be detected in LKB1-associated DNA by PCR. However, after LKB1 knockdown, no DNA segment corresponding to p21/WAF1 promoter was amplified taking LKB1 associated DNA as a template. Those results indicate that just like in tumor cells, LKB1 regulates the expression of p21/WAF1 by directly promoter-binding in hESCsMEF.
EMSA was then performed to test if nuclear transcription factor LKB1 bound to P21/WAF1 gene. As shown in Figure 2C, the nuclear extracts from hESCsMEF cells (untreated or transfection with mock siRNA, which contain the LKB1 protein) were incubated with oligonucleotide, results showed the significant shifts for p21 probe. After LKB1 knockdown, the band shift was blocked in hESCsMEF. In addition, hESCshAEC showed no binding activities was associated with the nuclear extract. These results indicate that p21 proteins bind to the LKB1's promoter. To test if the nuclear extract–probe complexes included LKB1 protein, antibody-based supershift assays were used by treating the complexes with LKB1 antibody before gel electrophoresis. As shown in Figure 2C, the LKB1 antibody caused significant supershift compared to the control sample containing no antibody. Consistent with previous studies, our findings further indicate that P21 is involved in LKB1 regulation in hESCsMEF.
LKB1 knockdown modifies the pluripotency of hESCs grown on MEF
To further determine the function of LKB1 in pluripotent controlling, we examined the expression of differentiation marker genes before and after RNAi in hESCs. As the original expression of LKB1 in hESCshAEC was low, the transfection of LKB1 specific siRNA almost did not influence the genes expression pattern (Fig. 3A). However, after LKB1 knockdown in hESCsMEF (Fig. 3B), the expression of Oct4, which is the stemness marker, was increased by about 4.5 times, although the genes related to early lineage-development such as Runx1, AFP, GATA, Brachyury, Sox17, and Nestin were downregulated by LKB1-RNAi (Fig. 3B).

Expression analysis of differentiation marker genes in hESCs after LKB1 knockdown by real-time PCR (18s rRNA was used as an internal control). (
These results suggest that maintenance of an undifferentiated self-renewal can be enhanced in hESCs grown on MEFs by interference with LKB1 knockdown, which is that LKB1 and by implication the p21/WAF1 pathway may be a negative regulator of the undifferentiated hESC phenotype maintained by this type of feeder.
Discussion
The LKB1/STK11 (serine/threonine kinase) tumor suppressor is established as an evolutionarily conserved regulator of cellular metabolism and cell growth (Shackelford and Shaw, 2009). More importantly, this function is not limited in somatic cells, but also in stem cells (Gan et al., 2010; Gurumurthy et al., 2010; Nakada et al., 2010). Depriving embryos of LKB1 leads to embryonic development deficiency in the mouse, indicating that this gene is critical for embryonic development (Jishage et al., 2002; Miyoshi et al., 2002). In this context, we demonstrate the role of LKB1 in pluripotency controlling of hESCs.
Since the first derivation of hESCs in 1998, the studies of hESCs biology have developed rapidly (Thomson et al., 1998). However, to provide more effective differentiated cells for regenerative medicine, the ESCs will be required continual maintenance of the undifferentiated state for prolonged periods in the culture system. The growth conditions and culture techniques might contribute to pluripotent state of hESCs (Hanna et al., 2010; Lengner e al., 2010). Here we showed that variable downregulation of ecto/meso/endodermal markers such as Nestin, brachyury, Runx1, AFP, GATA, and Sox17 occurred on hESCsMEF after Lkb1 RNA interference, whereas Oct4 expression was strikingly induced up to four- to five-fold (Fig. 3B). However, these differentiation marker genes did not have any change before or after Lkb1 RNA interference in hESCshAEC (Fig. 3A). These results probably reflects that LKB1 might regulated the differentiation potential in hESCs grown on MEF, and hAECs as a feeder layer to maintain hESCs might be more stable than MEF(Fig. 3B).
Although hESCs were derived from blastocyst inner cell mass, the in vitro cultured hESCsMEF represent the characteristics of in vivo implanted epiblast cells (Tesar et al., 2007). From morphology, the feature of hESCshAEC is closed to mESCs-like colonies. Our previous results demonstrated that hAECs might provide the proper microenvironment for hESCs to mimic the in vivo environment at preimplantation stage (Lai et al., 2010). Recently, Hanna et al. (2010) reported that human ES cells could be converted into a more immature state, which was achieved by ectopic induction of a few transcription factors, such as Oct4, Klf4, and Klf2 factors, combined with LIF and inhibitors of glycogen synthase kinase 3β (GSK3β) and mitogen-activated protein kinase (ERK1/2) pathway. These findings support the observation that distinct states of pluripotency can be specified by culture conditions; thus, the growth conditions need to be optimized to enhance the stability of naive human pluripotent state and to permanently stabilize this pluripotent state in the absence of genetic manipulations (Hanna et al., 2010). We employed CHIP assays and EMSA to test interaction of LKB1 with p21/WAF1 gene promoter by LKB1 RNA interference in human ES cell maintained on different feeder layers. As in somatic cells, which LKB1 function was shown to induce the transcription of p21/WAF1 gene (Shaw et al., 2004; Tiainen et al., 2002), we found that LKB1 directly regulates the p21/WAF1 gene through promoter-binding in hESCs growing on MEF. Because LKB1 expression could not be detected in hESCshAEC, we did not observe the regulation of these two genes in hESCs growing on hAECs. Thus, the tumor suppressor gene LKB1 expression may act as a negative regulator of a self-renewal in hESCs specifically grown on MEFs, which exhibit an inferior expression of pluripotency markers of stemness than hESC grown on hAEC. The absence of LKB1 mediated signaling is an important determinant of feeder cell-mediated support of hESC renewal.
Footnotes
Acknowledgments
We especially thank Dr. Paul A. De Sousa for critically reviewing this manuscript. This work was supported by grants from NSFC (National Natural Science Foundation of China, Project No 81070533) and Laboratory of Molecular Cell Biology, Chinese Academy of Science, Shanghai.
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.