
Abstract
Abstract
Production of transgenic animals via somatic cell nuclear transfer (SCNT) has been adapted worldwide, but this application is somewhat limited by its relatively low efficiency. In this study, we used handmade cloning (HMC) established previously to produce transgenic pigs that express the functional nematode fat-1 gene. Codon-optimized mfat-1 was inserted into eukaryotic expression vectors, which were transferred into primary swine donor cells. Reverse transcriptase PCR (RT-PCR), gas chromatography, and chromosome analyses were performed to select donor clones capable of converting n-6 into n-3 fatty acids. Blastocysts derived from the clones that lowered the n-6/n-3 ratio to approximately 1:1 were transferred surgically into the uteri of recipients for transgenic piglets. By HMC, 37% (n=558) of reconstructed embryos developed to the blastocyst stage after 7 days of culture in vitro, with an average cell number of 81±36 (n=14). Three recipients became pregnant after 408 day-6 blastocysts were transferred into four naturally cycling females, and a total of 14 live offspring were produced. The nematode mfat-1 effectively lowered the n-6/n-3 ratio in muscle and major organs of the transgenic pig. Our results will help to establish a reliable procedure and an efficient option in the production of transgenic animals.
Introduction
In an effort to improve the nutritional value of pork, we explored the possibility of introducing a gene facilitating omega-3 fatty acids synthesis into the swine genome. Omega-3 belongs to the family of n-3 polyunsaturated fatty acids (PUFAs), and has preventive and therapeutic effects on many illnesses, such as cardiovascular diseases (Gaibazzi and Ziacchi, 2004; Kris-Etherton et al., 2004; Mozaffarian et al., 2005; Simopoulos, 2008), arthritis (Ariza-Ariza et al., 1998; Kremer, 2000), cancer (Bougnoux, 1999; Cave, 1997; Rose and Connolly, 1999), and neuropathic diseases (Marszalek and Lodish, 2005). It is documented that n-3 PUFAs are not only essential for the normal function of the retina and brain (Uauy et al., 1996, 2000), but are also important as components of cell membrane phospholipids and precursors to the eicosanoid family of metabolites (Siddiqui et al., 2004).
A proper balance in the n-6/n-3 ratio is critical for maintaining normal body growth and development and for prevention of many clinical problems (Simopoulos, 2000). Unfortunately, humans and most mammals cannot synthesize n-3 PUFAs; the supply of n-3 fatty acids is totally dependent on dietary intake. Furthermore, the n-6 and n-3 PUFAs are not convertible in mammals due to a missing related converting enzyme (Kang, 2005). On the other hand, n-3 fatty acid desaturases in certain primary organisms can efficiently convert n-6 into n-3 PUFAs. Spychalla et al. (1997) cloned the fat-1 gene from the nematode Caenorhabditis elegans, and the gene was introduced into mammalian cells for production of n-3 PUFAs (Kang et al., 2001). Later, transgenic animals rich in n-3 fatty acids were cloned by the microinjection or SCNT method (Kang et al., 2004; Lai et al., 2006).
In the present study, we used HMC to produce transgenic pigs carrying the functional fat-1 gene from C. elegans. Codon-modified fat-1 (mfat-1) was inserted into eukaryotic expression vectors, which were transferred into primary swine donor cells. RT-PCR, gas chromatography, and chromosome analysis were performed to select recombinant donor clones capable of converting n-6 into n-3 fatty acids. Fourteen live piglets carrying the mfat-1 gene were produced, and the n-6/n-3 ratio was effectively lowered in the transgenic pigs examined. We have established a reliable HMC procedure to produce mfat-1 transgenic piglets with relative high efficiency.
Materials and Methods
Healthy female swine recipients were purchased commercially and kept in an experimental station dedicated for the transgenic swine research. Nuclear donor cells were obtained from primary cultures of porcine fetal fibroblasts (PFF) derived from a 40-day-old fetus of a Danish Landrace pig (Zhang et al., 2011). Fresh ovary tissues were collected from a contracted swine slaughter factory for isolation of the donor follicles. All chemical reagents were obtained from Sigma-Aldrich Chemical (St. Louis, MO, USA) except as otherwise indicated. Procedures for the animal experiments were approved by the Animal Care and Ethics Committee of the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences.
Construction of fat-1 expression vector
cDNA for the fat-1 gene was cloned from a laboratory stock of C. elegans, as described previously (Zhang et al., 2007). The coding region of the fat-1 gene (1209 bp) was optimized for mammalian expression (mfat-1) and placed into a pcDNA3.1 plasmid in frame to generate a CMV-mfat1-neo expression vector (Fig. 1). Another expression vector system (CAG-mfat1-neo) was constructed by replacing the cytomegalovirus (CMV) promoter with the CMV early enhancer and chicken β-actin promoter for the comparisons of expression efficiency (Fig. 1).
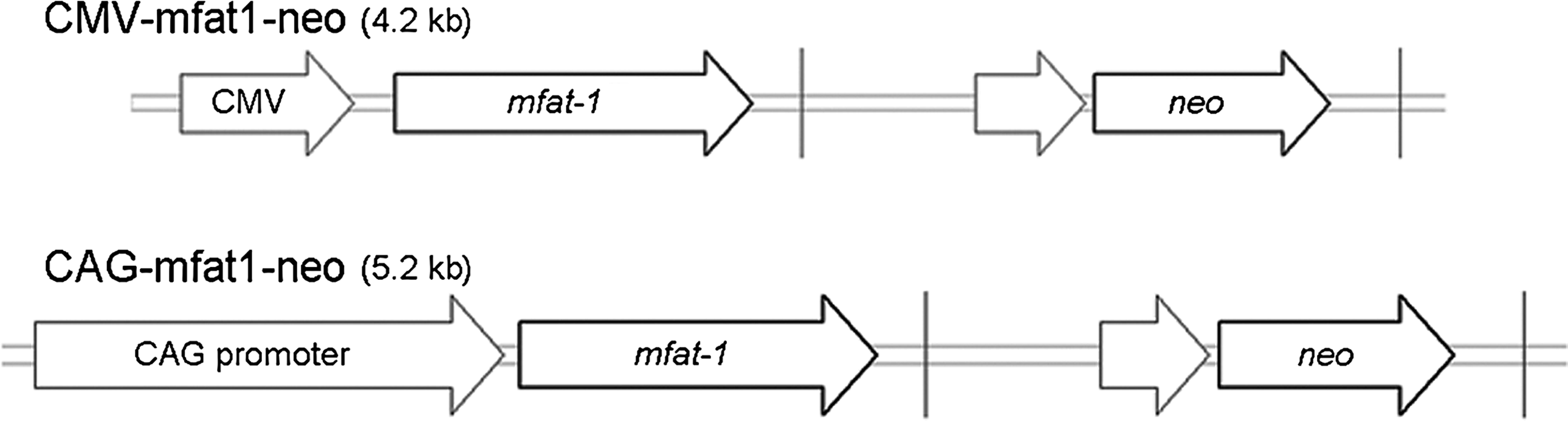
Schematic representation of n-3 fatty acid desaturase gene with linearized expression vectors. A codon-optimized mfat-1 gene was inserted into the pcDNA3.1 plasmid to generate CMV-mfat1-neo (CMV). CAG-mfat1-neo (CAG) was constructed by replacing the cytomegalovirus (CMV) promoter with the CMV early enhancer and chicken β-actin promoter.
Introducing the mfat-1 gene into PFF cells
PFF cells were grown in Dulbecco's modified Eagle's medium (DMEM; GIBCO, New York, NY, USA) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT, USA) (batch number NRE0007) at 38.5°C and 5% CO2. Approximately 1.5×105 cells were seeded into each well of a six-well plate 24 h prior to the transfection. For each well, 1.5 μg of linearized vector and 9 μL of FuGENE-6 (Roche Diagnostics, Indianapolis, IN, USA) were diluted with 91 μL of serum-free DMEM. After 25 min incubation at room temperature, the DNA–medium mixtures were added into 2 mL of cell culture medium. One day after the transfection, cells in each well were trypsinized and seeded onto one 10-cm culture dish (Becton Dickinson, Lincoln Park, NJ, USA) in medium containing 400 μg/mL Geneticin (Invitrogen). Drug selection was conducted for a period of 2 weeks with a medium change per 3 days. The Geneticin-resistant colonies were isolated and passaged at 80% confluency. Each colony was transferred into one well of a six-well plate to expand the cell population. The resulting clones were trypsinized and resuspended in medium containing 10% dimethyl sulfoxide (DMSO) at 80% confluency, then frozen at −80°C overnight and stored in liquid nitrogen for the HMC.
Analysis of mfat-1 expression in PFF
Geneticin-selected PFF cells were analyzed by PCR and RT-PCR for verification of the transgene construction. Genomic DNA of the cells was amplified for mfat-1 using the specific primer pairs (5′-ttatggtcgctcattcctca-3′ and 5′-ttcgattttacttggccttt-3′) under the following PCR conditions: Denaturation at 94°C for 5 min followed by 35 cycles of 94°C for 25 sec, 58°C for 20 sec, and 72°C for 1 min. For detection of mfat-1 transcripts, total cellular RNA was isolated from each candidate clone using TRIzol reagent (Invitrogen), and the cDNA was synthesized using RevertAid™ First Strand cDNA Synthesis Kit (MBI Fermentas, Ontario, Canada) according to instructions of the manufacturer. PCR was performed using the primer pairs and conditions described above.
The positive clones confirmed by PCR were subjected for the determination of mfat-1 mRNA expression by quantitative (q) real-time PCR, using a StepOnePlus™ Real-Time PCR machine (Applied Biosystems, Foster City, CA, USA). Sequence-specific primers for mfat-1 and GAPDH were designed, and Power SYBR® Green PCR Master Mix was used for all of the reactions at a 20-μL PCR volume (10 μL of 2× Master Mix, 2 μL of 20× primers, 6 μL of nuclease-free water, and 2 μL of cDNA sample). The PCR conditions were set as follows: 10 min at 95°C, 40 cycles of 95°C for 15 sec, 60°C for 25 sec, and 72°C 20 sec. Each qPCR experiment was run in triplicates.
Fatty acids analysis
The fatty acid compositions of total cellular lipids were analyzed for both of the recombinant and control clones using gas chromatography, as described previously (Kang and Wang, 2005). Fatty acid methyl esters were quantified using a fully automated 6890 Network GC System (Agilent Technologies, Palo Alto, CA, USA) with an Agilent J&W fused-silica DB-23 capillary column. The injector and detector were maintained at 250°C and 280°C, respectively. The oven program was maintained initially at 180°C for 10 min, then ramped to 200°C at 4°C/min and held for 15 min, and finally ramped to 230°C at 10°C/min and maintained for 6 min. Carrier gas-flow rate was maintained at a constant rate of 1.0 mL/min throughput. The peaks were identified by comparison with the internal fatty acid standards, and the area percentage for all of resolved peaks was analyzed using a GC ChemStation software (Agilent Technologies). Clones with a higher n-6 to n-3 turnover ratio were selected for the subsequent procedures of the HMC.
Karyotype analysis
The mfat-1–positive clones were incubated in a medium with 0.4 μg/mL demecolcine solution for 5 h, then trypsinized and incubated in 75 mM KCl at 37°C for 40 min, fixed in a fixation solution (methanol:acetic acid=3:1, v/v), and spread on microscopy slides for the morphological analysis. After Giemsa staining, the chromosome number and shape in each nucleus were examined at 1000× magnification. An average of 50 cells was examined for the selected positive clones, and the clones with any abnormal chromosome number or shape were eliminated.
Procedure outline of HMC
The ovaries collected at the slaughterhouse were kept in physiological saline solution within 2 h prior to the next step. Follicular fluid and cumulus–oocytes complexes (COCs) from follicles at 2- to 6-mm diameter were aspirated using a 14-gauge needle attached to a 10-mL vacuum collection tube. The compact COCs were selected and washed twice in HEPES-buffered tissue culture medium-199 (TCM-199; GIBCO). Each group of 50 COCs was matured in 400 μL of bicarbonate-buffered TCM-199 supplemented with 10% (v/v) cattle serum (CS), 10% (v/v) pig follicular fluid, 10 IU/mL antiequine chorionic gonadotropin (eCG), and 5 IU/mL human (h) CG in a CO2 incubator maintained at 38.5°C in a humidified atmosphere of 5% CO2.
The HMC was performed as described previously (Du et al., 2007). Briefly, at 42 h of in vitro maturation (IVM), the cumulus investment of the COCs was removed by repeated pipetting in 1 mg/mL hyaluronidase in HEPES-buffered TCM-199. Zonae pellucidae were partially digested with 3.3 mg/mL pronase solution dissolved in T33 (T for HEPES-buffered TCM-199 medium, 33% of the CS supplement). The oocytes with softened zonae pellucidae were lined up in T20 drops supplemented with 2.5 μg/mL cytochalasin B (CB), and were enucleated by oriented bisection with an ultrasharp microblade (AB Technology, Pullman, WA, USA) under a stereomicroscope. Less than half of the cytoplasm close to the polar body was removed manually from the remaining putative cytoplast. The positive transgenic cells were trypsinized and kept in T2.
Fusion was performed in two steps in which the second one included the initiation of activation (Du et al., 2005; Kragh et al., 2005). For the first step, half of the available cytoplasts were transferred into 1 mg/mL of phytohemagglutinin (PHA; ICN Pharmaceuticals, Australia) dissolved in T0, and then each one was quickly dropped over a single fibroblast. After attachment, cytoplast–fibroblast pairs were equilibrated in a fusion medium (0.3 M mannitol and 0.01% polyvinyl alcohol) for 10 sec. Using an alternating current of 0.08 kV/cm and 700 kHz (CF-150/B fusion machine; BLS, Budapest, Hungary), cell pairs were aligned to the wire of a fusion chamber (BTX Microslide 0.5-mm Fusion Chamber, model 450; BTX, SanDiego, CA, USA) with the somatic cells farthest from the wire, and then fused with a single direct current (D.C.) of 2.0 kV/cm for 9 μsec. After the pulse, cytoplast–fibroblast pairs were incubated in T10 drops to observe whether fusion had occurred. Approximately 1 h after the first fusion, each pair was fused with another cytoplast and activated simultaneously in activation medium (0.3 M mannitol, 0.1 mM MgSO4, 0.1 mM CaCl2, and 0.01% polyvinyl alcohol) by a single D.C. pulse of 0.85 kV/cm for 80 μsec. When fusion was observed in T10 drops, the reconstructed embryos were transferred into PZM-3 medium (Yoshioka et al., 2002) supplemented with 5 μg/mL CB and 10 μg/mL cycloheximide for 4 h at 38.5°C in 5% CO2, 5% O2, and 90% N2 with maximum humidity, then washed thoroughly with in vitro culture (IVC) medium before culture.
In vitro culture of reconstructed embryos
The reconstructed embryos were cultured in wells of a Nunc four-well dish in 400 μL PZM-3 medium supplemented with 4 mg/mL bovine serum albumin (BSA) and covered with oil. Zona-free embryos produced from the HMC were cultured in a modified well of the well (WOW) system (Feltrin et al., 2006; Vajta et al., 2000) at 38.5°C in 5% CO2, 5% O2, and 90% N2 with maximum humidity.
Cleavage and blastocyst formation of the reconstructed embryos were monitored during culture for 7 days. On day 7, a small portion of 7-day blastocysts were subjected to fixation and cell number counting for quality evaluation. Briefly, the embryos were stained with 20 μg/mL Hoechst 33342, and then incubated at 4°C for 24 h. The fixed and stained whole blastocysts were mounted and assessed for cell numbers using an epifluorescent microscope.
Embryo transfer and pregnancy diagnosis
The gilts that were at least 8 months old were used as the recipients. The blastocysts at day 5 to day 7 with clearly visible inner cell mass (ICM) produced from the HMC were surgically transferred to the uterine horns of naturally cycling gilts on 5 or 6 days of standing estrus. Pregnancies were diagnosed by ultrasonography on day 28 after the surgery and monitored every 2 weeks afterwards.
Screening of transgenic piglets
To detect the mfat-1 gene in cloned piglets, a small piece of tail tissue was collected from all piglets derived from the CAG-mfat1-neo transfected cells. Genomic DNA was extracted, and the presence of the transgene was verified by Southern blot or PCR, as described above. The tissue samples stored in liquid nitrogen were used for total RNA extraction, and the presence of target mfat-1 mRNA in the transgenic tissues was evaluated by RT-PCR.
Microsatellite analysis was performed for genomic DNA from each of the newborn piglets, the surrogate sow, and the donor fibroblasts. Ten polymorphic microsatellite loci (KS139, SO167, SW58, SW152, SW378, SW886, SW957, SW1987, SW1989, SW2116) located on different porcine chromosomes were amplified by three-color multiplex PCR, and the products were analyzed on a 3130XL Genetic Analyzer (Applied Biosystems) with GeneMapper ID Software v3.2 (Applied Biosystems).
For the Southern blot analysis, approximately 20 μg of genomic DNA was digested with BamHI for agarose gel electrophoresis. Blotting of the genomic DNA onto Hybond-N+ membrane (Amersham Pharmacia Biotech) and the hybridazation were performed as described (Ying et al., 2010). The probes were 32P -labeled PCR fragments of mfat-1 sequence, generated using the Random Prime Labeling System Redi Prime™II (GE Healthcare, Piscataway, NJ, USA).
To evaluate the activity of desaturase encoded by mfat-1 in transgenic pigs, one of the transgenic pigs was slaughtered at approximately 70 kg gross body weight for fatty acid analysis. The major organ and tissue samples (heart, liver, spleen, lung, kidney, tongue, ear, muscle, brain, testis) were collected from both transgenic and age-matched nontransgenic pigs, and the fatty acid analysis was performed as described above. The ratio of n-6/n-3 from each organ/tissue was used for comparison between the transgenic and the control pigs. Statistical analysis was performed using a two-tailed Student's t-test, and p values<0.05 were considered significant.
Results
Mfat-1 expression in PFF cells
We successfully introduced the mfat-1 gene into the genome of PFF donor cells. The PCR results showed that most of the geneticin-resistant cell clones carried the expected mfat-1 target gene (Fig. 2A). Quantitative PCR analysis of 28 positive clones of CAG-mfat1-neo and CMV-mfat1-neo constructs showed the level of mfat-1 mRNA expression in PFF cells varied significantly from clone to clone (Fig. 2B). Compared with the mRNA expression level normalized to the clone CAG1-4-4 (normalized value=1), much higher levels of mfat-1 mRNA expression were shown for the donor clones CAG3-2-2 and CAG1-3-6 (Fig. 2B). Accordingly, these two clones were selected for the subsequent assessment for conversion of n-6 fatty acids to n-3 fatty acids. Most of the cell clones derived from CMV-mfat1-neo expression vector showed a significantly lower level of mfat-1 mRNA (Fig. 2B), and the construct was removed from the system. The chromosomal analysis showed that the selected clones had a normal karyotype (see Fig. 2C for an example), indicating that these cell clones were competent as donor cells by the HMC.
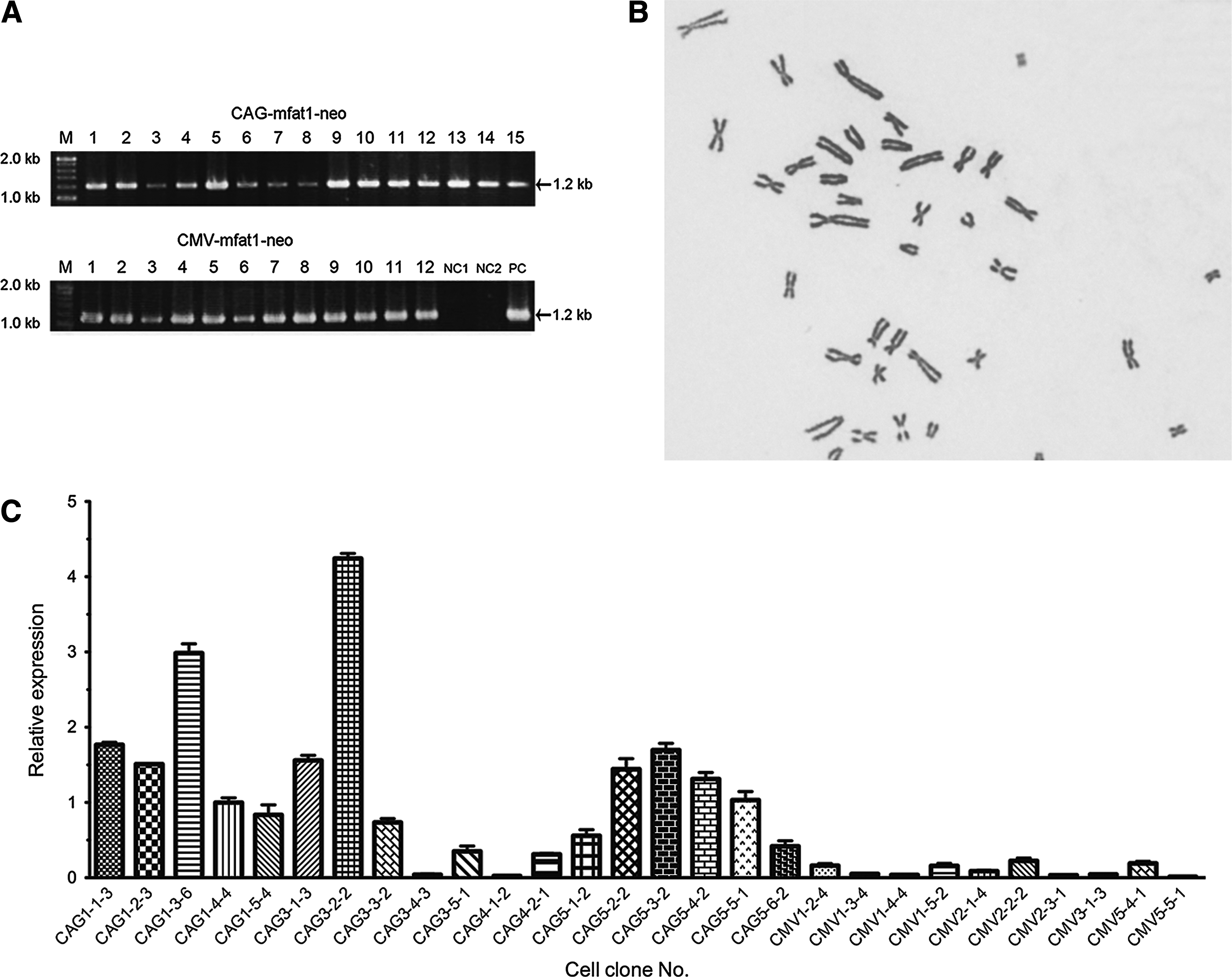
Identification and analysis of transgenic donor cell clones. (
Desaturase activity in PFF cells
Results of fatty acid composition analysis by the gas chromatography showed that the profiles of fatty acid in the transgenic PFF clones were remarkably different from those of the control cells (Fig. 3 and Table 1). In the cells expressing the mfat-1 gene, n-6 fatty acids, including 18:2n-6, 20:4n-6, and 22:4n-6, were converted to the corresponding n-3 fatty acids of 18:3n-3, 20:5n-3, and 22:5n-3, respectively (Fig. 3). Importantly, the ratio of n-6/n-3 was reduced from 11.48:1 in control cells to 1.16:1 (Table 1). In the control cells, such conversion peaks were not visible (Fig. 3).

Partial gas chromatograph traces showing the polyunsaturated fatty acid profiles of total cellular lipids from the CAG3-2-2 cells and the negative control cells. Note that the levels of n-6 polyunsaturated acids are lower, whereas n-3 fatty acids are abundant in the mfat-1 cells (right) as compared with the control cells (left), in which there is very little n-3 fatty acid (black arrows).
Fatty acid composition is presented as a percentage of the total cellular lipids from the control cells and mfat-1 cells (CAG3-2-2 and CAG1-3-6).
PUFA, polyunsaturated fatty acid.
Development of HMC embryos in vitro
Our results demonstrated that the HMC reconstructed oocytes with PFF donor cells have a higher rate of embryo development in vitro. For the HMC procedures, we collected a total of 4421 oocytes, of which 3410 (77%) reached the quality standard set for HMC and were utilized in subsequent operation (Table 2). Accordingly, a total of 1485 transgenic oocytes were reconstructed for embryo development and 558 of them successfully developed into the blastocyst stage of the embryo (Table 2). The averaged production efficiency for the blastocyst stage was 37.6±5.1% after IVC for 7 days, with a mean cell number of 81±36 per blastocyst (Fig. 4A, B).
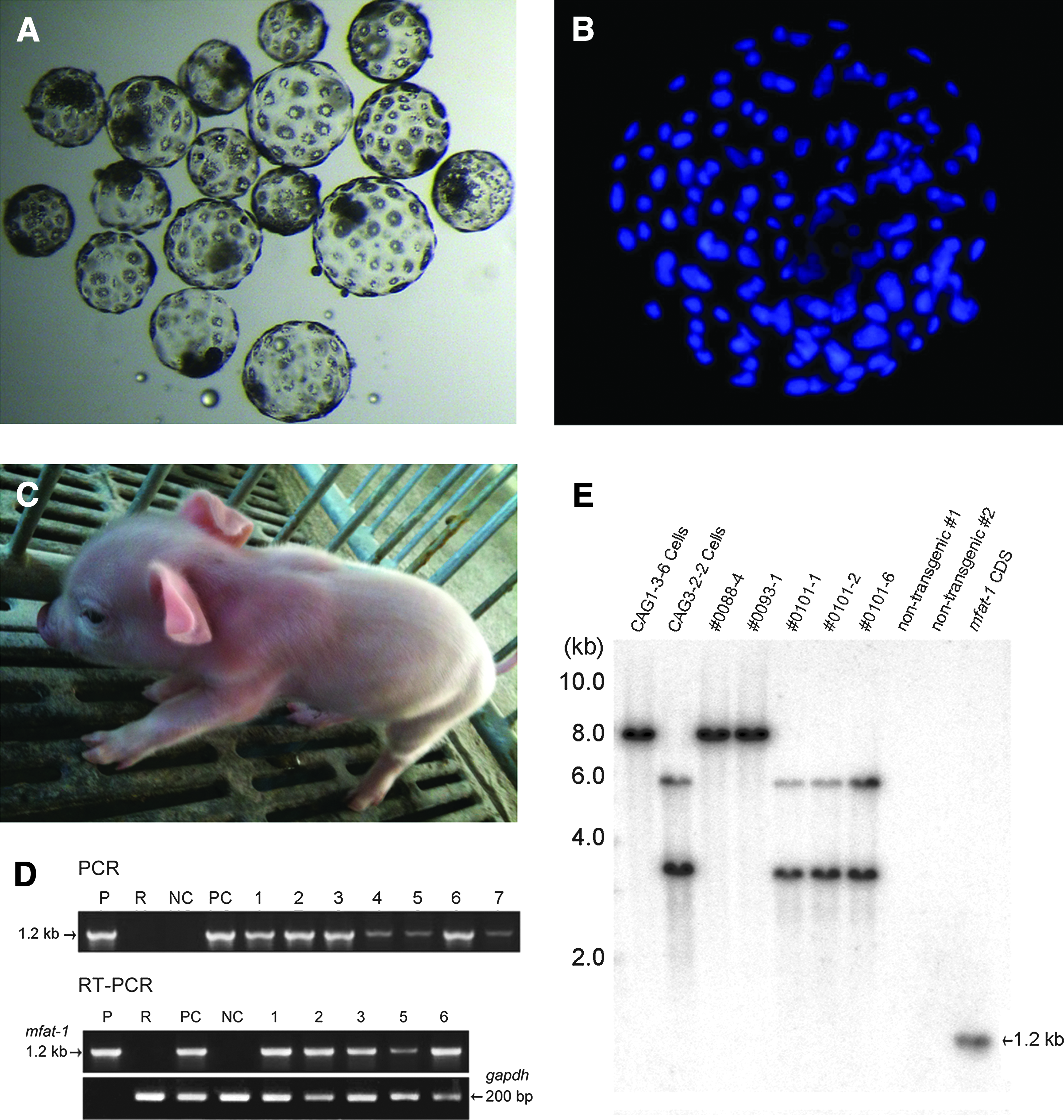
Production of transgenic blastocysts and piglets by handmade cloning (HMC). (
HMC, handmade cloning.
Production of mfat-1 transgenic piglets
A total of 408 blastocysts were transferred surgically to the uteri of four naturally cycling female recipients, with an average number of ∼100 blastocysts per recipient (Table 3). Following the transfer of blastocysts produced by the HMC, three recipients were pregnant and delivered a total of 16 live piglets (Fig. 4C). The average birth weight of the cloned piglets was 1.34 kg (range=0.50–1.90 kg). Most of the piglets seemed to be healthy and behaved normally. However, two of the seven piglets produced by recipient #0101 died a few hours after the birth, possibly due to the obvious lower body weights (0.50 kg and 0.80 kg, respectively) than average. Genetic analysis of all 14 piglets using 10 selected porcine microsatellite markers confirmed the identical genotype to those of the corresponding donor cells (data not shown). The presence of the mfat-1 gene was confirmed by PCR, RT-PCR, and Southern blotting (Fig. 4D, E). DNA sequencing of the PCR fragments confirmed that the target fragment was indeed the full-length cDNA of mfat-1.
HMC, handmade cloning.
Fatty acid analysis showed that the n-6/n-3 ratio in the transgenic pigs was significantly lowered in all of the tissues measured (Table 4). The n-3 fatty acids (α-linolenic acid, eicosapentaenoic acid, and docosahexaenoic acid) constituted approximately 4.2% of total muscle fat in the transgenic pig, much higher than levels in the nontransgenic controls, which was ∼0.7% on average. These results demonstrate that the mfat-1 gene was functional in the transgenic pig.
Ratio of n-6/n-3 fatty acid was calculated from n-6 fatty acids [linoleic acid (LA, 18:2n-6) and arachidonic acid (AA, 20:4n-6)] versus n-3 fatty acids α-linolenic acid (ALA, 18:3n-3), eicosapentaenoic acid (EPA, 20:5n-3), and docosahexaenoic acid (DHA, 22:6n-3)]. Each value represents the mean±standard deviation from three replicated sample measurements of each tissue.
Statistically significant as determined by Student's t-test (p<0.01).
Discussion
In this study, we generated 14 live transgenic piglets expressing the nematode mfat-1 gene without using the expensive micromanipulation instruments. The target gene was modified for optimal expression in mammalian cells, the presence and function of the transgene was verified in the donor cells before the nuclear transfer, and the modified mfat-1 gene in the transgenic pig lowered the n-6/n-3 ratio in all of the tissues investigated, indicating the functionality of mfat-1 gene in the swine genome. Although more studies are needed for answering questions related to the site of mfat-1 integration and the copy numbers in the swine genome, the techniques of HMC have been demonstrated again as a reliable alternative for the generation of transgenic animals (Kragh et al., 2009; Luo et al., 2011). Our work is a continuation of the previous efforts by other researchers. Similar work was done by Saeki et al. (2004), who successfully transferred a δ12 fatty acid desaturase gene from plant into the genome of swine by microinjection and increased the level of linoleic acid in the transgenic pig.
Our results illustrate the obvious advantage to evaluating the expression and function of the transgenic constructs and to preparing competent transgenic somatic cells prior to the SCNT. We performed RT-PCR to examine whether or not the transgene (mfat-1) was detectable in the genome of donor cells, and conducted gas chromatography analysis to compare the ratio of n-6/n-3 conversion between the transgenic and nontransgenic cell clones. This enabled us to select for the candidate clones that show a higher level of n-6/n-3 conversion rate, therefore increasing overall rate of success for the production of transgenic animal. Analysis of chromosomal number was also helpful in selecting competent transgenic donor cells before the SCNT, although our effort was limited to examine the chromosome number and shapes under the optical microscope.
In our study, the rate of blastocysts routinely reached 30–40% using the HMC method, with an average of 81 cells per blastocyst at day 6. Such efficiency is higher than that of the traditional nuclear transfer method, although the overall efficiencies of live piglets are similar (Petersen et al., 2008). Compared with previous studies that generated with the n-3 desaturase gene (Lai et al., 2006; Pan et al., 2010), our results for the birth rate and the efficiency of transgenic pigs (live transgenic offspring/born piglets) were higher (75.0%, 14/16 vs. 35.7%, 6/12; 70.0% 12/21). Using the HMC technique, two qualified technicians can routinely produce ∼100–140 reconstructed embryos in 2.5–3 h, generating enough blastocysts for the surgical transfer to a recipient animal (Schmidt et al., 2010).
Taken together, the work reported here helps to establish an efficient chain of analyses for selection of appropriate recombinant donor cells, and that HMC can be successfully applied for production of transgenic livestock with higher efficiency. This alternative technique will accelerate technology transfer and standardization and, eventually, might contribute to the widespread application of cloning.
Footnotes
Acknowledgments
We thank Lin Lin and Xin Xiong for technical assistance in microsatellite analysis. This work was supported by research grants from the National Basic Research Program of China (2004CB117500) and China Ministry of Agriculture (2009ZX08008-005B).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.