
Abstract
Abstract
Direct reprogramming of terminally differentiated cells to specify different cell types may allow somatic cells to be reprogrammed to an alternative, differentiated fate without intervening stem or progenitor cells. Recent studies have shown that the conversion of fibroblasts to other cell lines can be accomplished by the introduction of master regulator transcription factors. These findings have raised the question as to whether chemical molecules could replace transcription factor cocktails to directly alter defined somatic cell fate. Here, we demonstrate the generation of adipocytes directly from porcine embryonic fibroblasts (PEFs) using defined chemical molecules. Treatment with SB431542 and Thiazovivin, which are transforming growth factor-beta (TGF-β) and ROCK signaling pathway inhibitors, respectively, allowed PEFs to directly convert to fat-laden adipocytes. These induced adipocytes expressed multiple fat marker genes. We believe that these findings demonstrate that committed adipocytes can be directly reprogrammed from differentiated somatic cells using defined chemical molecules. The generation of adipocytes from nonadipogenic lineages has important implications for studies of adipogenesis, obesity modeling, and regenerative medicine. Additionally, these findings may enlighten a new method that direct reprogramming committed cell lines to other somatic cells using defined chemical molecules.
Introduction
Obesity is an epidemic that has become a major problem throughout the world in recent years (Roth et al., 2004). Adipose tissue is an important metabolic organ that is crucial for whole-body insulin sensitivity and energy homeostasis (Rosen and Spiegelman, 2006). Adipogenesis or adipocyte hyperplasia occurs throughout life in response to caloric intake that exceeds nutritional requirements (Prins and O'Rahilly, 1997). Being overweight or obese is associated with major health risks, such as cardiovascular disease, diabetes, nonalcoholic fatty liver disease, and cancer (Prins and O'Rahilly, 1997). Additionally, cachexia, a complex metabolic syndrome, results from the severe wasting of both fat and fat-free mass in certain disease states, such as cancer and immunodeficiency diseases. The conversion of adipocytes is therefore of great significance for studies on the mechanism of adipogenesis, the prevention of some fat-related diseases, and therapies for cachexia-accompanied diseases.
Some signaling pathways have been studied that play a vital role in regulating adipogenesis. Transforming growth factor-beta (TGF-β) regulates the differentiation program of a variety of cell types, including myoblasts, chondrocytes, osteoblasts, and adipocytes (Ballock et al., 1993; Choy et al., 2000; Janssens et al., 2005; Massague et al., 1986). TGF-β can inhibit the adipose differentiation of some preadipocyte cell lines (Ignotz and Massague, 1985; Petruschke et al., 1994), retain undifferentiated preadipocyte states (Ignotz and Massague, 1985; Sparks et al., 1992), reverse the adipocyte phenotype of some maturate adipocytes (Torti et al., 1989), and reduce the expression of differentiation markers of adipocytes (Petruschke et al., 1994). In TGF-β1 transgenic mice, both white and brown adipose tissue masses were reduced, and adipocyte differentiation was inhibited (Clouthier et al., 1997). Apart from TGF-β, Rho-associated kinase (ROCK) is also involved in adipogenesis. During adipogenesis, the actin cytoskeleton regulates the cell morphology of fibroblastic cells to round and fat-laden cells (Jaffe and Hall, 2005). ROCK regulates the reorganization of the actin cytoskeleton and cytoskeletal tension, and the inhibition of ROCK enhances adipogenesis (Noguchi et al., 2007).
In this study, we treated PEFs with the TGF-β signaling pathway inhibitor SB431542 and the ROCK signaling pathway inhibitor Thiazovivin to induce adipocytes. The induced adipocytes showed large lipid drops and the expression of the multiple fat marker genes. Moreover, we excluded the possibility that the induced adipocytes originated from meschenchemal stem cell (MSC) or progenitor cells and confirmed that the conversion occurred directly from the porcine embryonic fibroblasts (PEFs), without initial reversion to the pluripotent state. Our finding provides a new method for adipocyte differentiation that may result from adipogenesis. Importantly, our chemical molecule cocktail may serve as a new method with great potential for generating converted cell lines for regenerative therapies.
Materials and Methods
Ethics statement
Animal experiments were done in accordance with the guidelines on animal care and use established by the Jilin University Animal Care and Use Committee.
Cell culture
The PEFs were isolated from the 35-day-old fetuses of a 10-week-old Chinese miniature pig. After removing the head, tail, limbs, skeleton, and all internal organs, the fetuses were minced into pieces (1 mm3) and digested by Collagenase IV (Invitrogen, Carlsbad, CA) at 39°C for 4 h. Next, the cells were washed and seeded with 15 mL DMEM containing 15% fetal bovine serum (FBS) in 100-mm dishes. The culture medium for the fibroblasts was composed of DMEM supplemented with 10% FBS, 0.1 mM nonessential amino acids, 0.055 mM 2-mercaptoethanol, and 2 mM L-glutamine (Invitrogen). During induction, the PEFs were seeded at a density of 106 cells per dish in 60-mm dishes and were maintained in both chemical and hormonal induction medium at 39°C and 5% CO2. The chemical induction medium contained DMEM/F12 (Invitrogen), 15% Knockout Serum Replacement (Invitrogen), 0.1 mM nonessential amino acids (Invitrogen), 0.055 mM 2-mercaptoethanol (Invitrogen), 2 mM L-glutamine (Invitrogen), 2 μM Thiazovivin (Stemgent, San Diego, CA), and 0.5 μM SB431542 (Stemgent). The hormonal induction medium contained DMEM (Invitrogen), 15% FBS (Invitrogen), 0.1 mM nonessential amino acids (Invitrogen), 0.055 mM 2-mercaptoethanol (Invitrogen), 2 mM L-glutamine (Invitrogen), 5 μg/mL insulin (Sigma, St. Louis, MO), 1 μM dexamethasone (Sigma), and 0.5 mM isobutylmethylxanthine (Sigma).
Adipose conversion assay
The cells were washed twice with phosphate-buffered saline (PBS) (Invitrogen), fixed with 10% formaldehyde (Invitrogen) for 30 min, and then rinsed with 60% isopropanol (Sigma). After drying, 0.6% (w/v) Oil Red O (Sigma) solution (six parts 0.5% Oil Red O dye in isopropanol and four parts water) was used to stain the cells at room temperature for 30 min. Excess dye was quickly removed with 30% isopropyl alcohol, after which the cells were washed five times with water. To achieve relative quantitative measurements of lipid accumulation, the stained cells were covered with 2 mL isopropyl alcohol for 10 min, the extracted dye was collected, and its absorbance was measured at 510 nm.
Efficiency calculation
The total number of the adipocytes, defined as a round, fat-laden cell that can be stained with Oil Red O, was quantified after 20 days of induction. This number was determined in 10 randomly selected×20 visual fields from three independent experiments. The average per field was calculated and then was used to estimate the total number of the cells in the entire dish based on the known area of the×20 visual field and the entire dish. Then this number was divided by the number of plated PEFs before induction to get the efficiency of conversion.
Real-time reverse transcription-polymeras chain reaction (RT-PCR) analysis
The total RNA was isolated using Trizol (Invitrogen) from triplicate samples at five different time points during adipogenesis. The total RNA was purified with DNaseI (Fermentas, Hanover, MD) to remove genomic DNA contamination, and the RNA concentrations were measured using a NanoDrop-1000 (Thermo, Pittsburgh, PA). The cDNA was reversely transcribed using SuperScript III Reverse Transcriptase (Invitrogen). The expression of adipogenic transcription factors was determined by quantitative real-time RT-PCR using the SYBR Green PCR Master Mix (Invitrogen); all of the primers are listed in Table 1. β-Actin was used as an internal reference. The annealing temperatures of the primers were as follows: 60°C for β-actin and Adpioq, 62°C for PPARγ2, 55°C for C/EBPα, 57°C for C/EBPβ, and Fabp4, 58°C for C/EBPδ, 59°C for Lpl and Cfd, 53°C for Slc2a4. The cycle thresholds of all of the samples used for analysis were within the linear portion of the standard curve. The relative gene expression was calculated using the comparative Ct method with the formula 2−ΔΔCt (Livak and Schmittgen, 2001). The amplified PCR products were separated by 1.0% agarose gel electrophoresis and were then purified and confirmed by DNA sequencing.
Immunocytochemistry
The cells were washed twice with PBS, fixed with 4% formaldehyde for 30 min, and then covered with 0.1% Triton X-100 in PBS (Sigma) for 30 min. Five percent goat serum containing 0.1% Triton X-100 in PBS was added to block cells for 30 min at room temperature. The cells were incubated with 0.2% anti-PPARγ, C/EBPα, and C/EBPβ antibody (Sigma) containing 0.1% Triton X-100 in PBS for 3 h at 4°C. The cells were then treated with FITC goat antirabbit or antimouse antibodies (Sigma). The results were observed using a laser scanning confocal fluorescence microscope (Olympus, Melville, NY).
Western blot analysis
The cells were washed twice with cold PBS and harvested in NP40 Cell Lysis Buffer (Invitrogen) with phenylmethylsulfonyl fluoride (PMSF). After centrifugation, the supernatants were collected and quantitated by the QuantiPro™ BCA Assay Kit (Sigma). The protein samples were separated using SDS-PAGE and transferred electrophoretically onto polyvinylidene difluoride membranes (Amersham Biosciences, Piscataway, NJ). After blocking with 2% milk, the membranes were incubated with primary antibodies at 4°C overnight. The primary antibodies against PPARγ, C/EBPα, and C/EBPβ were from Sigma, and those against β-actin were from Abcam (Cambridge, MA). After washing with PBST, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (Sigma) for 2 h at room temperature. The signals were visualized by enhanced chemiluminescence (Perkin-Elmer, Norwalk, CT).
Statistical analysis
The results of lipid accumulation assay and real-time RT-PCR are expressed as the mean±SD (n=3). The statistical analysis was performed using Student's t-test for acomparison between two groups. The differences were considered significant if p<0.05.
Results and Discussion
The inhibition of the TGF-β and ROCK signaling pathway is sufficient for adipocyte induction
After culturing with SB431542 and Thiazovivin for 10 to 15 days, adipocytes were observed. During the subsequent induction, triacylglycerol lipid droplets accumulated and were stained with Oil Red O (Fig. 1A and B). The extend of adipose conversion, determined by the extraction of Oil Red O from stained cultures, was much higher in the induction using both SB431542 and Thiazovivin than when SB431542 was used alone (Fig. 1B and C). To determine the conversion efficiency, we estimated the total number of induced adipocytes in the entire dish on the day 20, and divided this number by the number of plated cells before induction to get a percentage of the starting population of cells that adopted adipocytes characteristics. The efficiency of using both inhibitors was 27.0%±5.7%, which is much higher than when using only SB431542 which is 17.3%±2.6%, and there was no adipocyte induction when using only Thiazovivin.

(
The expression of the fat marker genes including PPARγ2, C/EBPα, C/EBPβ, C/EBPδ, Lpl, Fabp4, Cfd, Slc2a4, and Adipoq was examined by real-time RT-RCR at different time points during the induction (5, 10, 15, and 20 days). As time passed, the expression of PPARγ, C/EBPα, Lpl, Fabp4, Cfd, Slc2a4, and Adipoq increased significantly, and the expression of C/EBPβ and C/EBPδ showed no remarkable differences (Fig. 2). Furthermore, the expression of these fat markers was much higher in the induction that utilized both inhibitors than it was when SB431542 was used alone, which further demonstrates that SB431542 is sufficient to convert PEFs to adipocytes and that Thiazovivin can promote the efficiency of the induction. The Western blot and immunofluorescence analysis also revealed the expression of PPARγ, C/EBPα, and C/EBPβ (Figs. 3–4). To confirm that SB431542 actually induced adipogenesis by regulating the TGF-β signaling pathway, we treated PEFs with SB431542 and TGF-β1 simultaneously and found no adipogenesis (data not shown). These results illustrate that PEFs can be converted into adipocytes by inhibiting the TGF-β signaling pathway and that the efficiency of adipogenesis can be promoted by downward modulation of the ROCK signaling pathway.

Real-time RT-PCR analysis of the mRNA levels of fat marker genes (PPARγ, C/EBPα, C/EBPβ, C/EBPδ, Lpl, Fabp4, Cfd, Slc2a4, and Adipoq) by induction with SB431542, combined SB431542 and Thiazovivin, and the hormonal cocktail. The expression levels of PPARγ, C/EBPα, Lpl, Fabp4, Cfd, Slc2a4, and Adipoq that were induced by combined SB431542 and Thiazovivin were significantly increased *p<0.05, **p<0.01 (Student's t-test) compared to the levels from SB431542 alone (n=3). The expression levels of C/EBPβ showed no remarkable change.
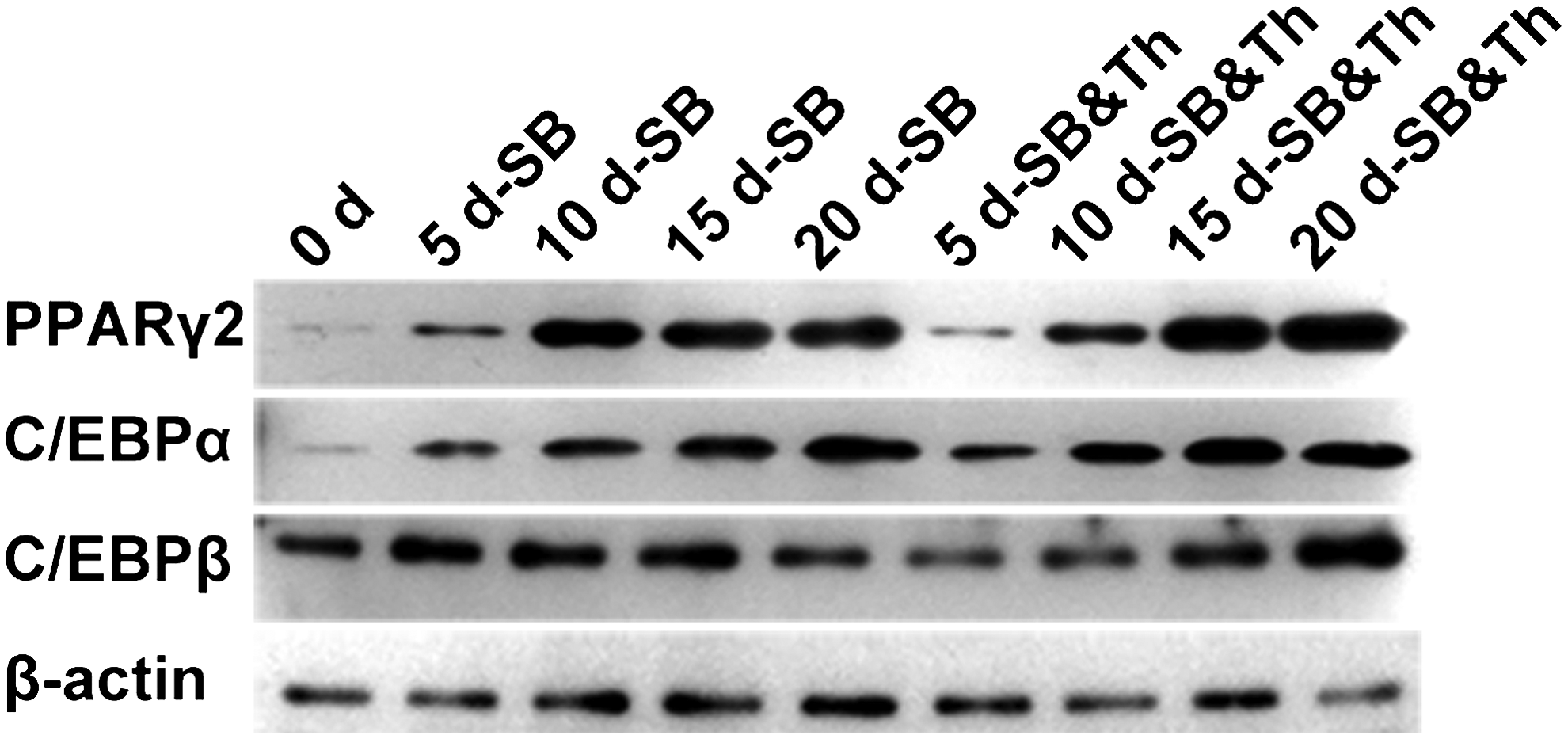
Western blot analysis shows the expression of PPARγ, C/EBPα, and C/EBPβ that was induced by SB431542 alone and by combined SB431542 and Thiazovivin treatment on days 5, 10, 15, and 20. β-Actin served as the loading control.
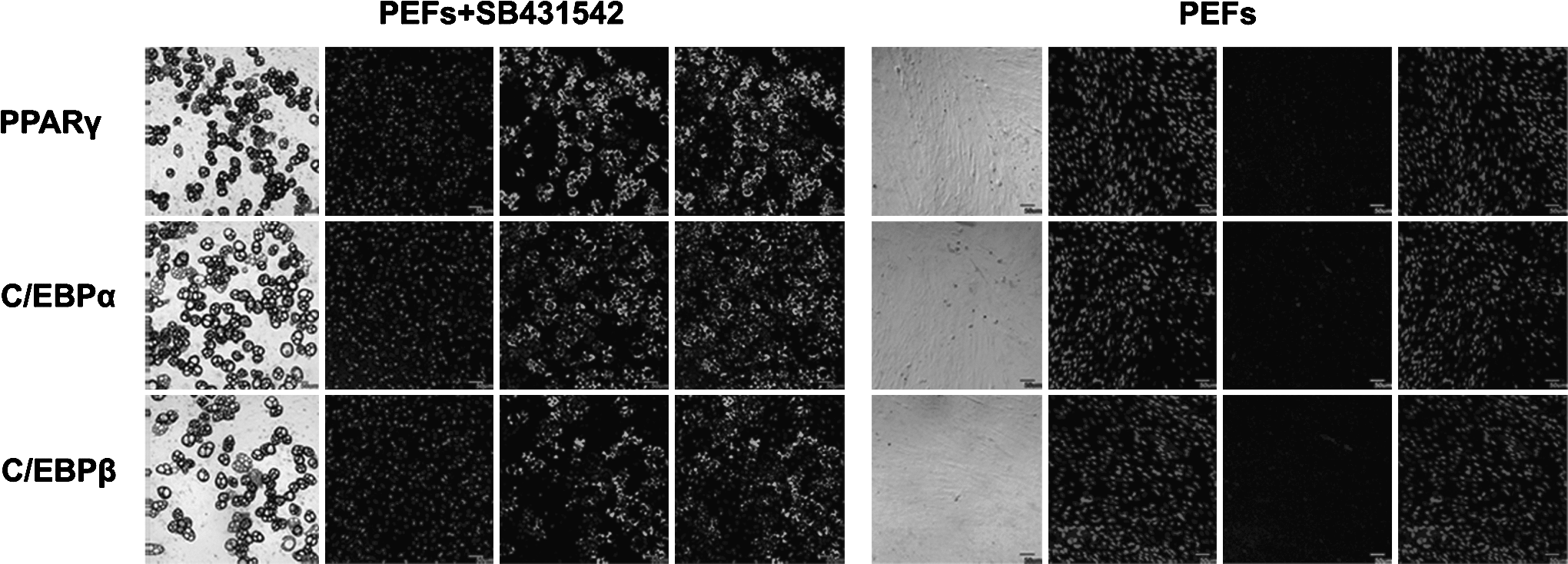
Immunofluorescence analysis of the expression of PPARγ, C/EBPα, and C/EBPβ in adipocytes induced by SB431542 on day 20. PEFs, which are untreated with inhibitors served as the control. Nuclei were stained with DAPI. Scale bar: 50 μm.
The interaction of the adipogenic transcription factors may provide insight into the molecular mechanism of adipogenesis. In response to hormonal stimulation, C/EBPβ and C/EBPδ are essential initiators of adipocyte differentiation, and they in turn activate the expression of PPARγ and C/EBPα (Fajas et al., 1998). Following adipocyte differentiation, the expression of C/EBPβ is downregulated, and C/EBPα, which exhibited increased expression induced by PPARγ activation, may also produce feedback that increases the expression of PPARγ. C/EBPα and C/EBPβ trigger growth arrest and initiate full adipocyte differentiation (Freytag and Geddes, 1992; Umek et al., 1991), whereas C/EBPα cooperates with PPARγ to activate the expression of adipocyte genes (Wu et al., 1999). During our adipocyte differentiation that used both SB431542 and Thiazovivin, the expression of C/EBPβ and C/EBPδ was nearly unchanged, and the expression of PPARγ and C/EBPα increased significantly (Fig. 2). The unchanged expression level of C/EBPβ and C/EBPδ may be in agreement with the observation that the inhibition of adipogenesis by TGF-β was accompanied by reduced mRNA levels of PPARγ and C/EBPα but not C/EBPβ and C/EBPδ (Choy et al., 2000). TGF-β targets the transcription factor cascade that is upstream of PPARγ, possibly by repressing the functions of C/EBPβ and δ (Choy et al., 2000). The enhancement of adipogenesis through the inhibition of the ROCK signaling pathway was likely due to the regulation of cell shape. Cell shape has been reported to regulate the commitment of human mesenchymal stem cells (hMSCs) by modulating endogenous RhoA, specifically via its effects on ROCK-mediated cytoskeletal tension (McBeath et al., 2004). A previous study has shown that expressing dominant-negative RhoA committed hMSCs to become adipocytes (McBeath et al., 2004), which provides a possible explanation for our finding that the RhoA effector, which is a ROCK inhibitor, increased the efficiency of adipogenesis.
The induced adipocytes were converted directly from PEFs
As it is known that MSCs and preadipocytes have the potential to generate adipocytes, we induced PEFs using a standard hormonal cocktail containing dexamethasone, isobutylmethylxanthine, and insulin to exclude the possibility that adipocyte progenitors existing in the isolated PEFs gave rise to adipocytes. After induction, there was no generation of adipocytes (Fig. 5A), little expression of PPARγ, and the expression levels of C/EBPα and C/EBPβ were lower and higher, respectively, than those found for treatment with SB431542 and Thiazovivin (Figs. 2 and 5B). The inability to induce high levels of PPARγ expression may be the most important reason that adipogenesis did not occur. These results excluded the possibility that the adipocytes arose from MSCs or preadipocytes that contaminated the PEFs.

(
To determine if the induced adipocytes passed through a stem-like state, we analyzed the expression of stem cell marker genes in four stages of adipocyte conversion. There was no expression of Nanog or Oct3/4 (Fig. 5C), which provided supporting evidence that the process of conversion to adipocytes bypassed the pluripotent state. In summary, our results indicate that the induced adipocytes were directly converted from PEFs.
In conclusions, our successful conversion of PEFs into adipocytes helps to improve the understanding of the mechanisms involved in adipogenesis and the prevention of diseases involved in adipogenesis. Future studies to improve the efficiency of the conversion are necessary to advance this technology for potential regenerative therapies. Our findings suggest that the utilization of signaling pathway inhibitors to convert cell lines can initiate sufficient expression of the endogenous genes that are involved in specific cell fate determination. In addition, cell shape, which changes dramatically during lineage reprogramming, can serve as a driving factor of this process. Furthermore, our chemical molecule-based method has more economical applications than the transgenic cocktail because it effectively eliminates any risk of genomic modification, it offers the safest method for altering the cell fates that have been described so far.
Footnotes
Acknowledgments
The authors express their gratitude to all the members of our laboratory for technical support and helpful discussion, and the staff at Embryo Engineering Center for technical assistance and taking care of the animals. This work was supported by China National Key Basic Research Program (973 Program, No. 2009CB941001, No. 2011CBA01003, and No. 2011CB944200).
Author Disclosure Statement
The authors declare that no conflicting financial interests exist.