
Abstract
Abstract
This study investigated the transdifferentiation of stem cells from human amnion tissue into functional acinar cells (ACs) using a co-culture system. Human amniotic epithelial cells (hAECs) were isolated from amnion tissue by mechanical mincing and enzymatic digestion. After primary culture, the phenotype of the cells was identified by flow cytometry (FCM) and immunocytochemical staining. hAECs were co-cultured with submandibular gland acinar cells of SD rats using a double-chamber system. The expression of α-amylase was determined by immunocytochemical method and fluorescent real-time quantitative reverse transcription polymerase chain reaction (RT-PCR) after induction for 1 and 2 weeks, respectively. Digestion with trypsin is an effective method for isolating hAECs from amnion tissue. These cells were positive for CD29 and CK19 and weakly positive for CD44 and α-amylase. Within 2 weeks, α-amylase in hAECs increased with induction time. The expression of α-amylase in hAECs was increased 3.38-fold after co-culturing for 1 week. This ratio increased to 6.6-fold, and these cells were positive for mucins, after co-culturing for 2 weeks. hAECs possess the potential to differentiate into ACs in vitro. They might be a stem cell resource for clinical applications of cell replacement therapy in salivary gland dysfunction diseases.
Introduction
Methods
Tissue collection and processing
This study was conducted in accordance with the Declaration of Helsinki and with approval from the Ethics Committee of ZunYi Medical College. Written informed consent was obtained from all participants. Fetal membranes were obtained from 3 healthy women by elective cesarean delivery for isolation and culture of ACs The animal use protocol was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of ZunYi Medical College. Submandibular gland ACs of SD rats were obtained from 8-day-old SD rats by the explant outgrowth technique without collagenase treatment, as previously described (Chen et al., 2005; Dimitriou et al., 2002). Eight-day-old SD rats were killed by suffocation. Using sterile techniques, the thin fascia covering the submandibular gland was carefully removed to expose the gland tissue, and then the submandibular gland was cut into small fragments of about 1 mm3 (Joraku et al., 2005). The tissue fragments were then cultured at 37°C in a 5% CO2 atmosphere in a humidified incubator with Dulbecco's modified Eagle's medium (DMEM)/F12 culture medium (Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco BRL, Grand Island, NY, USA ), 10%
Identification of ACs
ACs were identified by immunocytochemical analysis before co-culture. A portion of the cultured cells was plated on the cover glass for fixation with 4% paraformaldehyde for 30 min, washed with phosphate-buffered saline (PBS) three times, 0.3% Triton X-100 (Sigma, St. Louis, MO, USA) in PBS for 20 min, washed with PBS three times, and blocked with 1% bovine serum albumin (BSA) for 20 min. The primary antibody was monoclonal anti-α-amylase antibody (Santa Cruz), and cytokeratin-19 (CK19) and secondary antibodies (Dako Cytomation) were allowed to react with the cells for 30 min at 37°C to visualize the signal. Immunostained ACs were identified by microscopy, and the cultured cells were then prepared for co-culturing with hAECs.
Isolation and culture of hAECs
After informed written consent, human normal-term placentas were obtained after cesarean section or vaginal delivery. Samples were processed immediately at room temperature. First, the decidua parietalis was removed carefully by scraping. The amnion and chorion with adherent decidua were separated manually, and both membranes were washed three times in PBS with 100 U/mL penicillin and 100 g/mL streptomycin and cut into small pieces before enzymatic digestion.
Isolation of human amnion and chorion cells
Amnion fragments were incubated at 37°C for 10 min in PBS containing 0.05% trypsin followed by three rounds of exposure to 0. 05% trypsin in PBS at 37°C for 30 min. Cell suspensions were passed through a 200-μm cell strainer and collected by centrifugation (200×g for 10 min). The hAECs were cultured at 37°C in a 5% CO2 atmosphere in a humidified incubator with low-glucose (LG)-DMEM culture medium (Gibco BRL, Grand Island, NY, USA) supplemented with 10% FBS (Gibco BRL, Grand Island, NY, USA), 10%
Identification of hAECs
To evaluate cell-surface marker expression, cell suspensions were incubated for 20 min at 4°C with fluorescein isothiocyanate (FITC)- or phycoerythrin (PE)- conjugated isotype control or antibodies specific for human CD29 (clone MAR4) and CD44 (clone IM7). Samples were analyzed with a FACSCalibur and the CeIlQuest software. A portion of the cultured cells was plated on the cover glass for fixation with 4% paraformaldehyde for 30 min, washed with PBS three times, 0.3% Triton X-100 (Sigma, St. Louis, MO, USA) in PBS for 20 min, washed with PBS three times, and blocked with 1% BSA for 20 min. The primary antibody was monoclonal anti-vimentin antibody and CK19; secondary antibodies (Dako Cytomation) were allowed to react with the cells for 30 min at 37°C to visualize the signal. Immunostained hAECs were identified by microscopy, and the cultured cells were then prepared for co-culturing with ACs.
A portion of the cultured cells were plated on the cover glass, stained with Alcian Blue for 10 min, washed with distilled water for 5 min, staind with 1% periodate for 10 min, washed with distilled water for 5 min, stained with Schiff agent for 5 min, and washed with distilled water for 5 min. Alcain Blue–periodic acid Schiff (AB-PAS)–stained hAECs were identified by microscopy.
Co-culture system
hAECs were co-cultured with submandibular gland ACs in a double-chamber system by submandibular gland ACs on the upper inserts and hAECs at the lower culture plate area (Fig. 1). The ratio of cell density was 1:4 for hAECs and ACs. hAECs were treated with 1% EDTA and resuspended with DMEM/F12 culture medium at a density of 2×104 cells/well in a six-well plate (Costar, Corning Inc). Culture inserts (Costar, Corning Inc), each containing 8×104 ACs, were placed in four of the six wells, one insert for each well. The other two wells containing only hAECs served as controls. An insert membrane with a pore size of 0.4 μm in diameter was used to allow only the transmission of soluble factors and prevent direct interaction between hAECs and ACs.
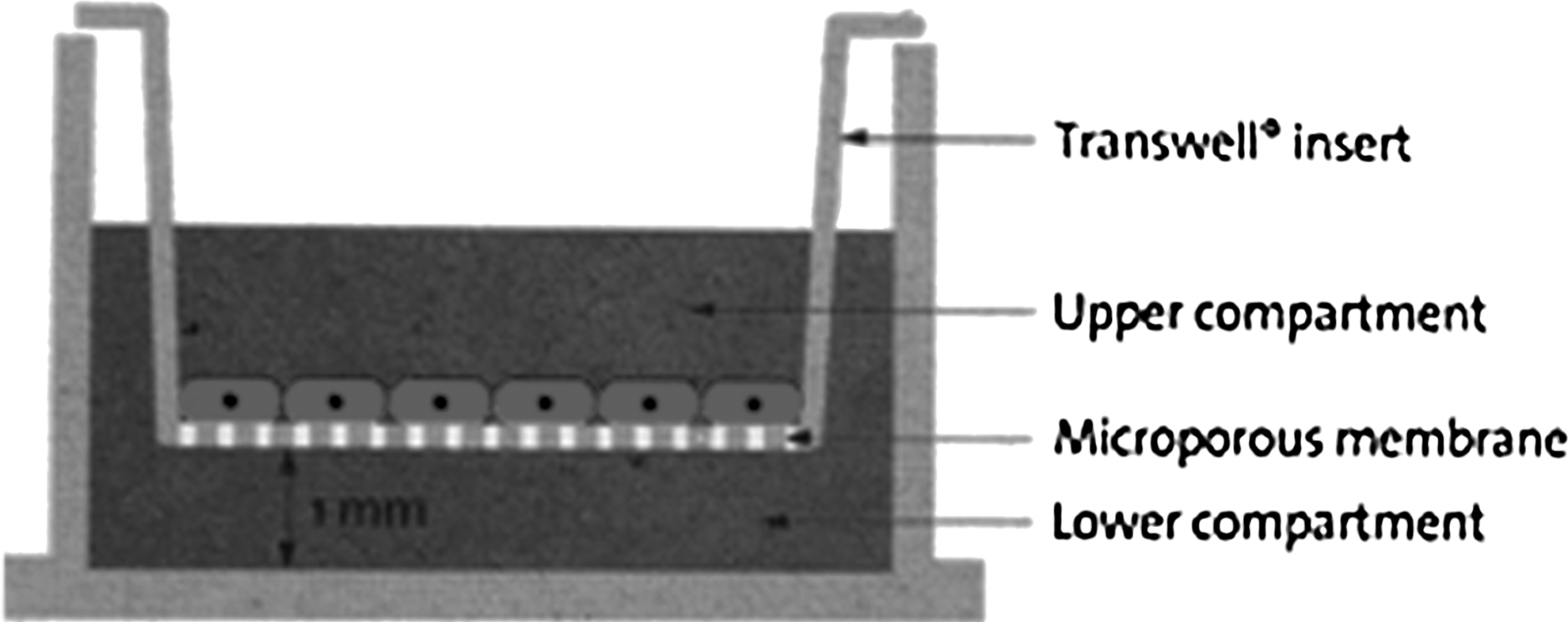
Illustration of the seeding of cells in the double-chamber co-culture system. The upper part of the co-culture system is comprised of six cell culture inserts with a 0.4-μm pore size to avoid direct cell-to-cell interaction. The lower part is a six-well plate. Co-culturing was performed by seeding ACs on the upper part and plating hAECs in the lower well. DMEM/F12 culture medium facilitated communication of the two parts via insert pores. Empty upper inserts that did not contain ACs served as controls.
Evaluation of the effects of the co-culture system
Morphologic changes of hAECs before and after transdifferentiation were investigated and compared with isolated ACs. Every week after co-culture, hAECs were subjected to immunocytochemistry to identify expression of the AC phenotype. After removing the medium and the inserts, the hAECs in the lower part of the culture plate were examined by immunocytochemical analysis for ACs as described previously.
Fluorescent real-time quantitative RT-PCR
Total RNA was extracted from retrieved PGA scaffolds and cultured cells using an RNase kit (Takara Biotechnology Co., Dalian, China) according to the manufacturer's instruction. PCR was performed using the primers for the human gene encoding α-amylase (forward, 5′-CATAAGGATGAGCAAGCATAAATC-3′, reverse, 5′-TCGCAAGTGGAATGGAGAG-3′), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (forward, 5′-GCACCGTCAAGGCTGAGAAC-3′, reverse, 5′-TGGTGAAGACGCCAGTGG-3′). After removal of the cells from the porous membrane with lysis buffer from the RNeasy kit, RNA was extracted and treated with RNase-free DNase according to the manufacturer's protocol (Takara Biotechnology Co., Dalian, China). One microgram of total RNA was reverse transcribed into cDNA using the First Strand cDNA Synthesis Kit (Takara Biotechnology Co., Dalian, China), according to the manufacturer's instructions. PCR reaction mixtures contained cDNA template, PCR primer, and SYBR Premix Ex Taq™ in a final volume of 20 μL. PCR reactions were performed on an C1000 thermal cycler (Bio-Rad, USA). Thermal cycling parameters were: 10 sec at 95°C, followed by 40 cycles of 5 sec at 95°C and 20 sec at 60.2°C, and 84 cycles of 10 sec at 55°C.
Statistical analysis
Relative quantification of gene expression between multiple samples was achieved by normalization against two endogenous controls GAPDH, as recommended by Willems et al. (2006), using the ΔΔCt method of quantification. The relative amount of mRNA was calculated as 2−ΔΔCt and real-time quantitative reverse transcription polymerase chain reaction (RT-PCR) results were statistically analyzed by one-way analysis of variance (ANOVA). Data are presented as mean±standard error of the mean (SEM), and statistical significance was defined as p<0.05.
Results
Identification of ACs
The explant outgrowth technique and DMEM/F12 medium were used to culture submandibular gland ACs from 8-day-old SD rats. Cells could be seen around the tissue after 5 days in primary culture. Cells were subcultured for 4 days to about 90% confluence to obtain sufficient cells. The expression of α-amylase and CK19 in the cells was determined by immunocytochemical staining. Production of α-amylase is one of the most characteristic functions of ACs.
Identification of hAECs
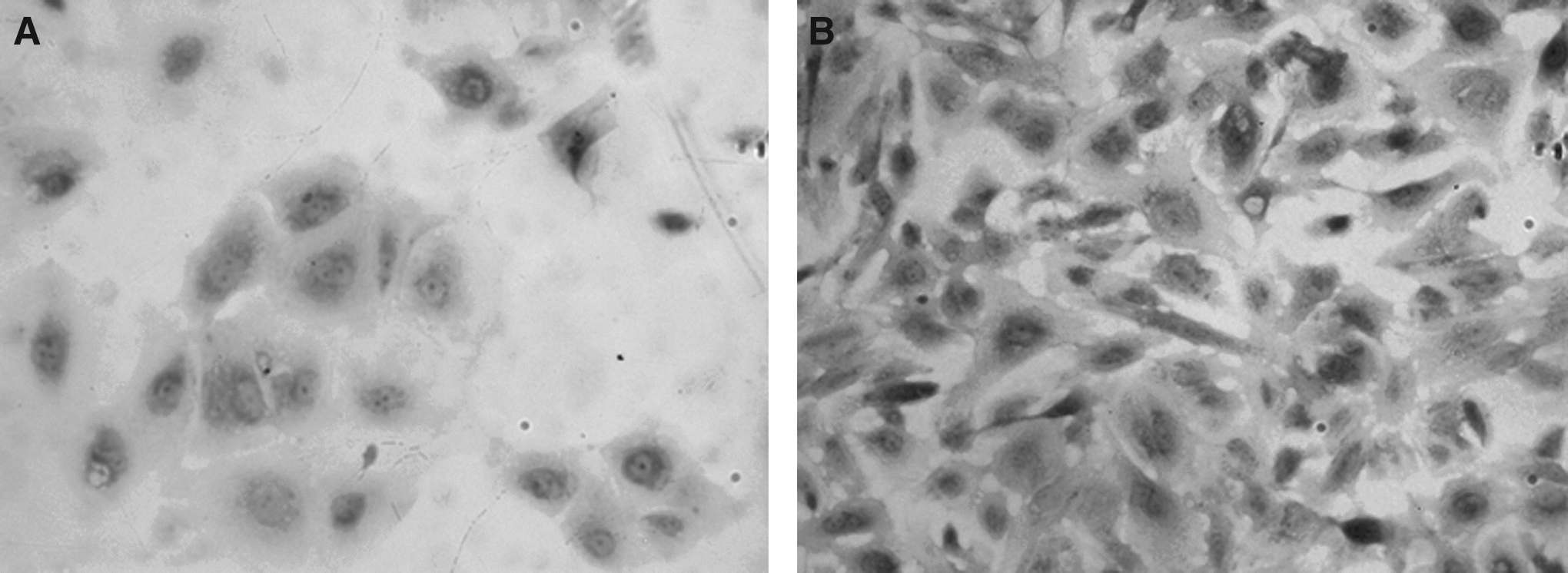
After mechanical separation of amnion and chorion membranes and treatment with trypsin, about 5.0×107 hAECs were obtained from each amnion tissue. Cell activity was >90%, and surface antigens expressed by hAECs were analyzed by flow cytometry (Fig. 2). These cells within the sixth passages were positive for CD29 and weakly positive for CD44. hAECs from human term fetal membranes within the sixth passages were characterized by immunocytochemistry staining (Figs. 3 and 4) and were positive for epithelial CK19 and weakly positive for α-amylase. However, proliferation was slowed down and the cell body changed after the sixth passage.

Flow cytometric analysis of hAECs. (
Immunofluorescence staining for CK19 in hAECs. Magnification, 400×.

(
Evaluation of the effects of the co-culture system
Immunocytochemical and fluorescent real-time quantitative RT-PCR analysis were performed every week after the start of co-culture to determine the expression of the AC functional protein α-amylase to determine whether hAECs could transdifferentiate into ACs. Expression of α-amylase was increased in hAECs after 2 weeks of co-culture. None of the control group experiments, which involved hAECs alone, showed expression of α-amylase. Figure 4 shows the expression of α-amylase, indicating the transdifferentiation of hAECs after co-culture for 2 weeks. At weekly intervals after the start of co-culture, three wells of hAECs were examined and then three additional wells were examined under the same conditions. hAECs were weakly positive for α-amylase before co-culture.
Differentiation of hAECs after co-culture
The mRNA levels of α-amylase were studied at different time intervals, Within 2 weeks, α-amylase in hAECs increased with induction time (Fig. 5). There was a significant difference in expression of α-amylase every week after co-culture between the experimental and control groups. The mucins in the hAECs cytoplasm were evaluated with AB-PAS staining. Before transdifferentiation, mucins were weakly positive. After 2 weeks of co-culture, hAECs strongly expressed mucins in their cytoplasm (Fig. 6).

(
(
Morphologic investigation of cells
Before transdifferentiation, the hAECs were either of orbicular-ovate or trianglular shape, arranged in slabstone configuration. After co-culture the secretory granules in cytoplasmic increased and were like those of ACs (Fig. 7).

(
Discussion
Radiation therapy is considered an effective local treatment for head and neck cancer. This treatment is accompanied by hyposalivation, a condition that has a negative impact on patients' quality of life. Pilocapine and amifostine can enhance salivary secretion, or serve as a radioprotector of salivary glands for patients treated with cancer therapies (Burlage et al., 2009; Haddad et al., 2009; Jensen et al., 2010). Gene transfer is also used to restore or prevent radiation-induced damage to salivary glands and their hypofunction (Gao et al., 2011; Palanyandi et al., 2011). Until now, there is no effective preventive treatment or therapy for head and neck cancer patients whose salivary gland function has been lost through radiological treatment. Thus, development of an effective implantable tissue engineering salivary gland organoid is becoming urgent. The problem of seeding cells is the key issue for salivary gland tissue engineering. Because ACs derived from primary cultures have a finite life span, other cell sources should be used. One solution is the use of stem cells (Aframian and Palmon, 2008). Adult salivary gland stem cells are considered promising candidates for cell therapy and tissue regeneration after irreversible salivary gland damage has occurred in head and neck cancer patients undergoing radiotherapy (Coppes and Stokman, 2011; Feng et al. 2009; Lombaert et al., 2011). Bone marrow–derived cells (BMDCs) were reported to rescue salivary gland function (Sumita et al., 2011).
Recently, researchers have reported that hAECs have multipotent differentiation ability and may serve as a source of cell transplantation therapy. hAECs possess considerable advantageous characteristics. They can differentiate into all three germ layers, such as neurocyte, hepatocyte, and islet cell (Kakishita et al., 2000; Scaggiante et al., 1987; Takashima et al., 2004; Wei et al., 2003); they have low immunogenicity and antiinflammatory functions (Rebmann et al., 1999; Rooney and Morgan, 1992); and sacrifice of human embryos is not required for their isolation, thus avoiding the controversy of using human embryonic stem cells. Therefore, hAECs derived from the discarded amnion after parturition are expected to serve as an attractive material in the field of regenerative medicine (Toda et al., 2007).
In this study, we successfully established a model of transdifferentiation of hAECs into ACs by using a double-chamber system. We used an explant outgrowth procedure without tissue digestion to successfully maintain the growth and phenotypic expression of primary cultures of SD rat submandibular gland ACs. We examined the phenotype of hAECs. hAECs were positive for CD29 and CK19 and weakly positive for CD44 and α-amylase. These results show that the separation of target cells in this experiment results in a high purity of hAECs, and these cells have the stem cell phenotype of certain CDs.
With immunocytochemical methods, we found that α-amylase in hAECs increased with a longer time of co-culture. Fluorescent real-time quantitative RT-PCR reaction showed the expression of α-amylase in hAECs was increased 3.38-fold after co-culturing for 1 week. This ratio increased to 6.6-fold after co-culturing for 2 weeks. Expression of α-amylase in the cytoplasm increased and was more easily observed with time. Mucins were strongly expressed in the hAECs cytoplasm after co-culture with ACs (Fig. 6). The morphologic changes in the hAECs after co-culturing with ACs also showed increased similarity to ACs as shown in Figure 7. Thus, we speculated that a cytokine from the submandibular gland cells leads to hAECs differentiation.
hAECs can differentiate into ACs in vitro in the presence of SD rat salivary gland cells by using a double-chamber system. Co-culture of BMSCs with respiratory epithelial cells (Le Visage et al., 2004), cardiomyocytes (Garbade et al., 2005), and myocytes (Xu et al., 2004) by direct cell-to-cell contact has been shown to induce transdifferentiation, but not without direct cell contact. Rangappa et al. (2003) indicated that, in addition to soluble signaling molecules, direct cell-to-cell contact is obligatory in relaying the external cues of the microenvironment controlling the differentiation of adult stem cells to cardiomyocytes. These studies indicated that an indirect method would be more difficult than a direct-contact co-culture system.
Our study has demonstrated the possibility of transdifferentiation of hAECs into ACs. These results showed the effect of an indirect co-culture method for transdifferentiation of hAECs. However, further studies are needed to test the function of these acinar-like cells and their phenotypic similarity to ACs, which will determine their potential to serve as an unlimited cell source for the development of an artificial salivary gland for the treatment of patients with salivary hypofunction. (Lin and Lee, 2007). In fact, we had transplanted hAECs by direct intraglandular injection into the damaged SD rat salivary gland. These hAECs were still alive after 3 weeks (data not shown).
In conclusion, we found that hACEs possess the ability to transdifferentiate into acinar-like cells in the presence of appropriate environmental conditions. Further studies are needed to investigate the signal transduction and mechanisms involved in the transdifferentiation of hAECs into ACs. Studies are also needed to overcome limitations of the general application of hAECs and this co-culture system. In addition, potential and functional effects of hAECs differentiation to ACs in the body (in situ) still need further analysis.
Footnotes
Acknowledgments
The authors would like to thank the Key Laboratory of Cell Engineering of Guizhou Province, Affiliated Hospital of Zunyi Medical College. Funding was provided by the Research Foundation of GuiZhou Science and Technology Department [SY (2011) 3016] and the research Foundation of GuiZhou province office of education [(2009) 0112].
Author Disclosure Statement
No competing financial interests exist.